Identification of global alternative splicing and sexspecific splicing via comparative transcriptome analysis of gonads of Chinese tongue sole (Cynoglossus semilaevis)
2022-06-07YiFangLuQianLiuKaiQiangLiuHongYanWangChengHuaLiQianWangChangWeiShao
Yi-Fang Lu, Qian Liu, Kai-Qiang Liu, Hong-Yan Wang, Cheng-Hua Li, Qian Wang,*, Chang-Wei Shao,*
1 School of Marine Sciences, Ningbo University, Ningbo, Zhejiang 315211, China
2 Key Lab of Sustainable Development of Marine Fisheries, Ministry of Agriculture, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao, Shandong 266071, China
3 Laboratory for Marine Fisheries Science and Food Production Processes, Pilot National Laboratory for Marine Science and Technology,Qingdao, Shandong 266000, China
4 College of Fisheries and Life Science, Shanghai Ocean University, Shanghai 201306, China
ABSTRACT Chinese tongue sole (Cynoglossus semilaevis) is an economically important marine fish species with a ZZ/ZW sex determination mechanism, which can be influenced by temperature.Alternative splicing (AS)is an important mechanism regulating the expression of genes related to sex determination and gonadal differentiation, but has rarely been reported in fish.In this study, to explore the molecular regulatory mechanisms of sex determination and gonadal differentiation, we combined isoform and RNA sequencing (Iso-Seq and RNA-Seq) to perform transcriptome profiling of male and female gonads in C.semilaevis.In total, 81 883 and 32 341 full-length transcripts were obtained in males and females,respectively.A total of 8 279 AS genes were identified, including 2 639 genes showing differential AS (DAS) between males and females.Many intersecting DAS genes and differentially expressed genes (DEGs) were enriched in the meiotic cell cycle pathway, and genes related to gonadal differentiation, such as esrrb and wt1a, were found to have sex-specific isoforms.Thus, this study revealed AS events in the gonadal transcriptomes of male and female C.semilaevis, described the characteristics of active transcription in the testes, and identified candidate genes for studying the regulatory mechanisms of AS during gonadal differentiation.
Keywords: Gonadal differentiation; Alternative splicing; Sex-specific splicing; Cynoglossus semilaevis; Iso-Seq; RNA-Seq
INTRODUCTION
Alternative splicing (AS) is a ubiquitous mechanism for regulating gene expression that allows the generation of multiple mRNAs from a single gene.During this process,RNA-binding proteins (RBPs) can influence the recognition efficiency of splicing complexes at splicing sites and thus modulate AS (Witten & Ule, 2011).Types of AS are categorized as skipped exons (SEs) (particularly exon removal), mutually exclusive exons (MXEs) (only one of two exons is retained after splicing), alternative 5’/3’ splice sites(A5SSs, A3SSs) (affecting boundaries between introns and exons), and retained introns (RIs) (Baralle & Giudice, 2017).AS can increase the diversity of mRNA and protein isoforms and alter the expression of gene isoforms.For example,90%-95% of human genes undergo AS, and approximately 37% of genes generate multiple protein isoforms (Kim et al.,2014; Pan et al., 2008).AS can also change translation reading frames to regulate gene expression, known as nonsense-mediated decay (NMD) (Isken & Maquat, 2008).In addition, the A5SSs and A3SSs of mRNA untranslated regions (UTRs) can affect the stability, localization, and translation efficiency of mRNAs (Licatalosi & Darnell, 2010).Thus, due to its ability to enrich different types of mRNA,further functional analysis of AS is necessary.
AS plays an important role in sexual development and gonadal differentiation in teleosts.In Chinese tongue sole(Cynoglossus semilaevis), the ovarian germ-cell-specificvasagene has three isoforms in the gonad, includingvas-s, which shows sexually dimorphic expression during early gonadal differentiation (Wang et al., 2014).Another germ-cell-specific gene, factor in the germline alpha (figla), expresses two isoforms,figla_tv1andfigla_tv2, which are separately expressed in the oocytes of female and germ cells of pseudomale testes (Li et al., 2016).Knockdown offigla_tv2in pseudomale testes significantly up-regulates the expression of two steroid hormone-coding genes, suggesting the involvement offigla_tv2in spermatogenesis via regulation of steroid hormone synthesis (Li et al., 2016).In Nile tilapia,SRY-box containing gene 30 (sox30) is expressed exclusively in the gonads, and its four AS-generated isoforms are expressed at different stages of gonadal differentiation, thus showing clear sexual dimorphism (Han et al., 2010).In seabream, four progesterone receptor gene (pgr) variants are co-expressed in the ovary, among which two isoforms show differential expression under gonadotropin and estrogen stimulation, suggesting that ovarian progestin responsiveness may be regulated by AS ofpgrmRNA during early oogenesis(Zapater et al., 2013).Thus, AS appears to be a universal regulatory mechanism of sexual development and gonadal differentiation in teleosts.
The Chinese tongue sole is an economically important and sexually dimorphic marine species, with females showing faster growth than males.As such, sex determination and gonadal differentiation in this species have become important areas of research.However, while genes related to sex determination and gonadal differentiation have been investigated, few studies have explored the mechanisms underlying differences in gene expression in female and male gonads, especially AS of key genes during gonadal differentiation.To perform a comparative transcriptomic analysis of expression and AS regulation between Chinese tongue sole females and males, we combined PacBio isoform sequencing (Iso-Seq) and short-read RNA sequencing (RNASeq) to generate a comprehensive transcript dataset of the gonads after differentiation.Abundant differentially expressed genes (DEGs) and genes with differential alternative splicing(DAS) were obtained.Based on enrichment analysis, DAS in DEGs was related to mRNA splicing and germ cell development.Our results provide a foundation and potential candidate genes for further research on the mechanisms underlying differential expression between the ovary and testis ofC.semilaevis.
MATERIALS AND METHODS
Ethics statement
All animal procedures followed the principles of the Guide for the Care and Use of Laboratory Animals at the Chinese Academy of Fishery Sciences and were approved by the Institutional Animal Care and Use Committee (IACUC) of the Yellow Sea Fisheries Research Institute (CAFS) (Qingdao,China)(Approval No.: YSFRI-2022016).
Sample collection of C.semilaevis
In this study, a batch of healthy Chinese tongue sole (at 6 months post-fertilization (mpf)) was sampled from Laizhou Mingbo Co., Ltd.(Yantai, Shandong, China).The gonads were collected and immediately frozen in liquid nitrogen and stored at -80 °C for RNA extraction.The caudal fins were collected and stored in ethanol for genetic sex identification.
Histological observation
The collected gonadal tissues were fixed in 4%paraformaldehyde (PFA) at 4 °C overnight, then soaked in 10 mmol/L phosphate-buffered saline (PBS) (Solarbio Science, China) for 1 h.The fixed samples were dehydrated through a series of graded ethanol concentrations, embedded in paraffin blocks, and cut into 6 μm sections.The sections were fixed on slides and stained with hematoxylin-eosin(Solarbio Science, China).The slides were photographed with a Leica DM4000 B light microscope (Leica Microsystems,Germany).
Genetic sex identification
Genomic DNA was extracted from the fins using the phenolchloroform method.Genetic sex was identified by polymerase chain reaction (PCR) amplification of sex-specific simple sequence repeat (SSR) markers (Liu et al., 2014).
T otal RNA extraction
Total RNA was isolated from each gonad sample using TRIzol reagent (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions.RNA sample quality was measured with an Agilent 2 100 Bioanalyzer (Agilent, USA).
RNA-Seq and data processing
The libraries of three females (F1-F3) and three males(M1-M3) were sequenced on the BGISEQ-500 platform.Raw reads were filtered using SOAPnuke v1.4.0 (Chen et al., 2018)and Trimmomatic v0.36 (Bolger et al., 2014) with default parameters to remove adapters, low-quality reads with more than 5% unknown nucleotides, and reads with more than 20%low-quality bases.The filtered clean data were subsequently mapped to theC.semilaevisgenome (NCBI Cse_v1.0) with HISAT2 (v2.1.0) (Kim et al., 2019) and aligned with the reference transcript sequence using Bowtie2 (v2.2.5)(Langmead & Salzberg, 2012) to remove ribosomal RNA(rRNA) sequences and annotate genes.
Full-length transcriptome sequencing and data processing
Four full-length transcriptome libraries were constructed by using RNA samples from the gonads of each two females and two males.Total RNA was enriched with oligo (dT) magnetic beads and then reverse transcribed to produce cDNA using a SMARTer™ PCR cDNA Synthesis Kit (Takara Bio, USA).The full-length cDNA was amplified by PCR, and the BluePippin™Size Selection System (Sage Science, USA) was then used for size selection to generate four libraries (insert size of 1-10 kb).After selection, the full-length cDNA was amplified again and end repaired, and SMRT sequencing adaptors were ligated to the cDNA to produce the SMRT bell libraries.The libraries were quantified using a Qubit RNA BR Assay Kit(Thermo Fisher Scientific, USA).The library insert size was checked with an Agilent 2 100 Bioanalyzer, and sequencing primers and polymerase were bound to the SMRT template in appropriate proportions according to the PacBio calculator sequencing results obtained from the PacBio Sequel platform(Pacific Biosciences, USA).We first performed raw read quality control and filtration, removing reads with low quality and short length, and then generated circular consensus sequences (CCSs) by filtering based on number of full passes greater than zero and accuracy greater than 0.75.The CCSs were classified and clustered using SMRT Link (v5.1.0)software supported by Pacific Biosciences.For the clustered CCSs, we performed alignment to theC.semilaevisgenome(NCBI Cse_v1.0) using GMAP software (v2017.06.20), and the final transcriptome sequences were generated using TOFU (v1.0) to remove redundant sequences.We annotated the full-length transcriptome and identified novel transcripts by matching the alignment results to the genome annotation file with MatchAnnot (v1.0), and then used TransDecoder (v3.0.0)to predict the open reading frames (ORFs) of full-length transcripts.
Expression analysis
The sequences aligned by Bowtie2 were quantified by RSEM(v2.2.5), and gene expression levels were normalized using the fragments per kilobase million (FPKM) method to eliminate the influence of sequencing depth and gene length.Differential expression analysis of normalized gene expression was performed using DEseq2 (v1.30.1) (Love et al., 2014),with fold-change≥2 and adjustedP(q-value)<0.05 indicating significant DEGs.
DAS analysis
We utilized the lr2rmats pipeline (v0.1) to integrate SMRT long-read and RNA-Seq short-read data, thus generating a new gene annotation file.Clean RNA-Seq reads were then aligned to the genome using STAR (v2.5.3a) (Dobin et al.,2013), resulting in an alignment file for each sample.The newly generated annotation and alignment files for each sample were fed into rMATS (v4.1.0) (Shen et al., 2014) for AS analysis, whereby short reads were used to compare differences in reads per kilobase million (RPKM) in specific regions of the transcripts derived from each gene.For each AS event, rMATS calculated percentage of exon inclusion(IncLevel) for each sample across the biological triplicates and detected differential IncLevel (IncLevelDifference) between the two sexes.The DAS events were screened and categorized using summary.py in rMATS based on a IncLevelDifference absolute value greater than 0.1 and false discovery rate (FDR)of less than 0.05.The splicing event categories included SEs,A5SSs, A3SSs, MXEs, and RIs.
Functional analysis of differential AS genes
Gene Ontology (GO) enrichment analysis was performed using Metascape (https://metascape.org).Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was performed based on annotation results from the alignment ofC.semilaevisprotein sequences in the KEGG database(https://www.genome.jp/kegg/).
Visualization of AS events
Sashimi plots of AS events were generated using rmats2sashimiplot (v2.0.3).RNA-Seq and full-length transcript structures were viewed and visualized in Integrative Genomics Viewer (IGV) (v2.8.13) (Robinson et al., 2011).
Gene expression validation by quantitative real-time PCR(qRT-PCR)
To verify splicing regulator expression based on RNA-Seq data, eight candidate genes potentially involved in gonadal differentiation were selected.Primers for the selected genes were designed based on the longest mRNA sequences generated from theC.semilaevisgenome sequence(Supplementary Table S1).The RNA-Seq samples of each group were employed for cDNA synthesis using a PrimeScript RT Reagent Kit with gDNA Eraser (TaKaRa, Japan).qRTPCR was performed using a QuantiNova SYBR Green PCR Kit (Qiagen, Germany).Total qRT-PCR volume (20 μL)contained 10 μL of 2×SYBR Green PCR Master Mix, 2 μL of QN ROX reference dye, forward and reverse primers (each at 0.7 μmol/L), and 1 μL of cDNA.PCR amplification was performed using a StepOnePlus Real-Time PCR system(Thermo Fisher Scientific, USA) under the following conditions: 95 °C for 2 min, followed by 40 cycles at 95 °C for 5 s and 60 °C for 10 s, with final collection of the fluorescence signal of the dissolution curve.The PCR assays were performed in triplicate.Theβ-actingene was used as an internal reference gene.The relative expression levels of eight genes were measured using the 2-∆∆Ctequation.Thet-test was used for assessment of significance, andP<0.05 was considered statistically significant.
RESULTS
Histological observations of gonadal differentiation
At 6 mpf, the ovarian cavity and seminal lobules were observed in the ovary and testis, respectively, indicating completion of gonadal differentiation.In the ovary, oocytes were orderly distributed along the ovarian lobules, with few oogonia (Figure 1A).In the testis, many seminal vesicles were wrapped around the testis periphery and filled with sperm.Germ cells, including spermatogonia, spermatocytes, and spermatozoa, were observed in the seminal lobules(Figure 1B).
AS events in gonads
To fully characterize AS in the gonads, we performed Iso-Seq and RNA-Seq to obtain full-length transcripts and their relative expression levels.We identified 32 341 and 81 883 transcripts in the ovary and testis, with mean transcript lengths of 1 670 and 2 301 bp, respectively.In total, 9 957 and 14 258 genes were annotated, respectively.Among those transcripts, we identified 1 321 (ovary) and 4 038 (testis) novel transcripts,which were shorter in length than known transcripts in the gonads (Supplementary Figure S1A, B).Furthermore, 6 648 and 11 088 genes showed AS in the ovary and testis,respectively (isoform number≥2) (Figure 2A).The ratio of male to female genes increased with isoform number, indicating that genes expressed in the testis had more isoforms than those in the ovary, which was especially true for genes with more than 10 isoforms (1 878) (Figure 2A).AS events were predicted using short RNA-Seq reads, resulting in 21 325 AS events in the gonads (Figure 2B).The SE, A5SS, A3SS, MXE,and RI splicing types accounted for 70.47%, 6.07%, 7.98%,4.98%, and 10.49% of all splicing events, respectively.
Enrichment analysis of DAS genes between females and males
In total, 8 279 AS genes were identified in females and males.After filtering (|IncLevelDifference|>0.1 and FDR<0.05), 2 639 genes showing DAS were obtained (Supplementary Table S2).The SE, A5SS, A3SS, MXE, and RI slicing typesaccounted for 48.16%, 8.83%, 8.98%, 6.97%, and 27.06% of AS events, respectively (Figure 3A).KEGG enrichment analysis of these genes was performed (Supplementary Tables S3-S7), and a Venn diagram of the top 30 pathways of each type showed that the spliceosome and mRNA surveillance pathways were shared among the five types of DAS genes (Figure 3B).We also identified several other enriched pathways involved in gametogenesis, such as oocyte meiosis, basal transcription factors, and cell cycle.SEs were mainly found in the oocyte meiosis pathway.Structural maintenance of chromosomes protein 1B (LOC103379931,smc1b) andpgr-like (LOC103391230) contained sex-specific exons (Supplementary Figure S2A, B).In the basal transcription factor and cell cycle pathways, the RIs of these genes mainly occurred in males.In addition, TATA boxbinding protein-like 2 (LOC103381809,tbpl2), transcription initiation factor TFIID subunit 9 (LOC103395620,taf9),transcription initiation factor IIA subunit 1 (gtf2a1), and TFIIH subunit XPD (ercc2) exhibited male-specific introns(Supplementary Figure S2C, D; Figure 3C, D).In the cell cycle pathway, male-specific RIs were also found in meiosis-specific stromal antigen 3 (stag3), cyclin-dependent kinase 7 (cdk7),and stromal antigen 2 (LOC103395545,stag2) (Figure 3E, F;Supplementary Figure S2E).

Figure 1 Light microscope images of C.semilaevis gonads at 6 mpf
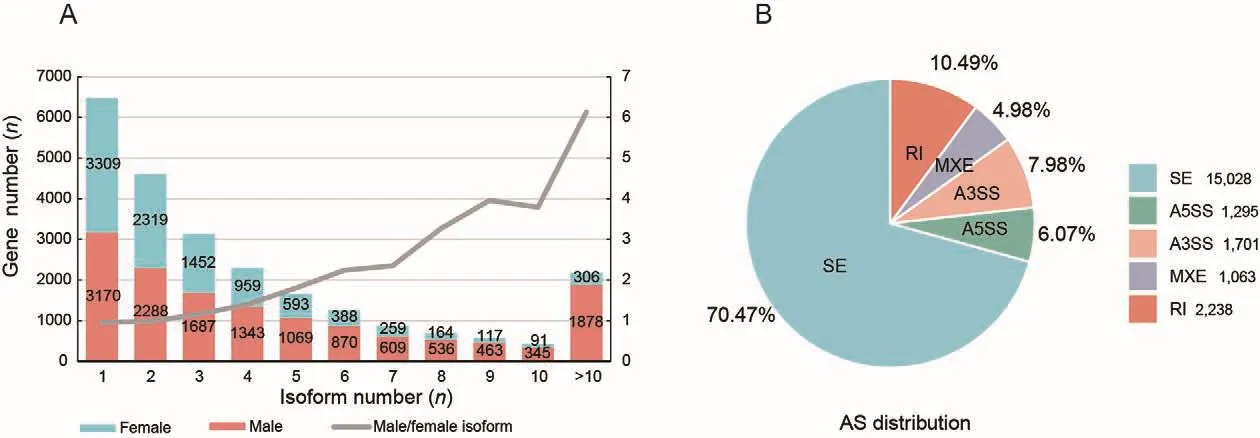
Figure 2 Number and type distribution of AS events in gonads of C.semilaevis
Effect of AS on gene expression
There were 5 077 intersecting genes between the 8 279 AS genes and 13 444 DEGs (Figure 4A).We counted the number of DEGs showing AS and found that SEs were the dominant AS type (Figure 4B).The proportions of differentially expressed AS genes containing SEs, A5SSs, A3SSs, MXEs,and RIs were 61.53%, 57.75%, 61.55%, 61.53%, and 57.22%,respectively (Figure 4C).More genes were up-regulated in females than in males in all AS categories, except RIs(Figure 4C).We also found that the expression levels of genes showing AS events were higher than the expression levels of genes not showing AS events (Figure 4D).
AS regulates expression of genes related to gonadal differentiation
In total, 1 112 DEGs were screened from 2 639 DAS genes(Figure 5A).The network of enriched GO terms is shown in Figure 5B.Meiotic cell cycle-related GO terms, such as meiotic cell cycle process and homologous recombination,were enriched (Supplementary Table S8).
A total of 25 genes related to gonadal differentiation were identified (Figure 5C).We focused on two genes in particular:i.e., steroid hormone receptor ERR2 (esrrb), which encodes a protein similar to the estrogen receptor that functions as a transcription factor (Festuccia et al., 2018), and WT1 transcription factor a (wt1a), which is involved in urogenital development (Perner et al., 2007).Our results showed thatesrrbwas significantly up-regulated in the ovary compared with the testis (q-value<0.01), and a female-specific 4th exon existed, which was missing in males (Figure 5D).In the testis,wt1awas markedly expressed (q-value<0.01) and an intron between the 9th and 10th exons was specifically retained(Figure 5E).
Role of splicing regulators in gonadal differentiation
Based on the enrichment results, we focused on the other significantly enriched GO term, i.e., regulation of RNA splicing(Figure 5B), which included 24 splicing regulator genes.These genes showed different expression patterns between females and males (Figure 6A).Certain genes were up-regulated in females, including DEAH-box helicase 35 (dhx35), DEAH-box helicase 8-like (LOC103381857,dhx8-like), serine/argininerich splicing factor 1a (LOC103378294,srsf1a), and splicing factor U2AF 35 kDa subunit-like (LOC103391595,u2af1-like),while certain other genes were up-regulated in males,including transformer 2 beta homolog (tra2b), DEAD-box helicase 5 (LOC103383782,ddx5), transformer 2 alpha homolog (tra2a), and serine/arginine-rich splicing factor 1b(LOC103395123,srsf1b).
To validate the RNA-Seq data, the expression levels of eight genes, i.e.,dhx35,dhx8-like,srsf1a,u2af1-like,tra2b,ddx5,tra2a, andsrsf1b,were measured by qRT-PCR(Figure 6B).Their expression trend patterns were consistent with the sequencing data.

Figure 3 Number distribution and enriched pathway analysis of genes showing DAS in female and male gonads of C.semilaevis
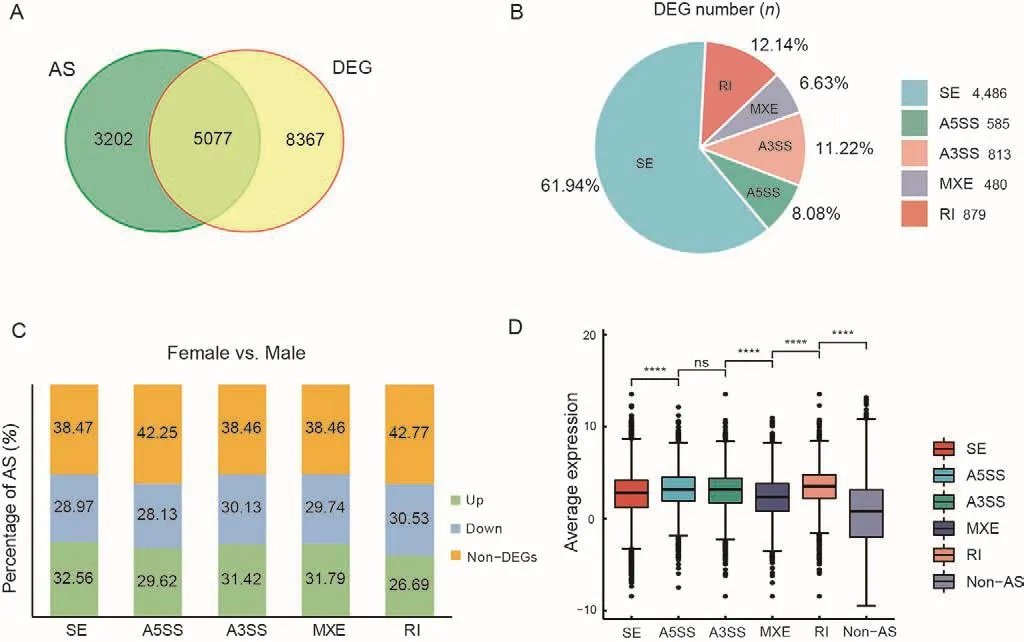
Figure 4 Analysis of AS and DEG association in female and male gonads of C.semilaevis

Figure 5 Function and expression analysis of intersecting DAS genes and DEGs
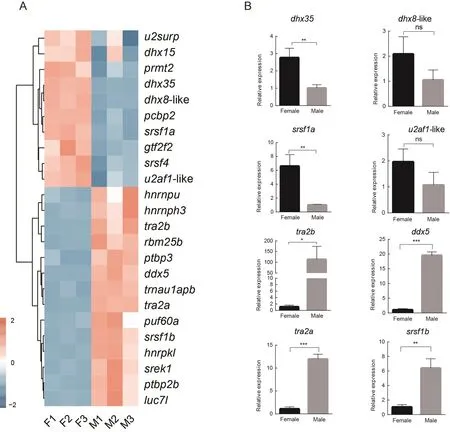
Figure 6 Gene expression and validation of splicing regulators in gonads of C.semilaevis
DISCUSSION
While studies have implicated the involvement of AS in sexdetermination and gonadal differentiation in many species(Carreira-Rosario et al., 2016; Cesari et al., 2020; Gómez-Redondo et al., 2021; Kuravsky et al., 2011; Wang et al.,2014; Yu et al., 2021), knowledge on the regulatory mechanisms of AS in gonadal differentiation remains limited.Cynoglossus semilaevisis an economically important and sexually dimorphic fish species, with females considered more valuable than males in the aquaculture industry due to their faster growth.Research on sexual development and gonadal differentiation inC.semilaeviswill provide theoretical support for the development of a monosex fish breeding industry.In this study, with the help of high-throughput sequencing technology, we explored the different regulatory mechanisms involved in gonadal differentiation inC.semilaevismediated by AS.
AS events were more extensive in C.semilaevis testis
Based on Iso-Seq analysis, the number of full-length transcripts in the testis was 2.53 times higher than that in the ovary (81 883/32 341), and more genes were transcribed in the testis than in the ovary, indicating extensive and active gene transcription in the testis, as reported in mammals and birds (Soumillon et al., 2013).Here, many mRNAs were transcribed and novel transcripts identified in theC.semilaevistestes, consistent with previous study showing that the permissive environment of chromatin in the testis facilitates new genes, especially in spermatocytes and sperm cells(Kaessmann, 2010).In addition to the active transcription of genes in the testis, the average number of transcripts per gene in the testis (5.74) was higher than that in the ovary(3.25), thus reflecting more active AS in the testis.RNA-Seq showed more up-regulated genes in the testis than ovary,which may be related to the active transcription occurring in this organ.In mammals, RIs and SEs are the most common types of AS (Braunschweig et al., 2014; Kalsotra & Cooper,2011).We identified 21 325 transcripts showing AS, with SEs and RIs also found to be the most common splicing types inC.semilaevis(accounting for 80.96%), thus suggesting conservation of AS type in mammals and teleosts.
Differential SEs and RIs were found in gametogenesisrelated pathways
We compared DAS between the testis and ovary, with SEs and RIs again found to be the dominant DAS types (75.22%).Genes containing SEs, such assmc1bandpgr-like, were enriched in the oocyte meiosis pathway.Thepgrgene is an important ovulation gene in preovulation follicular granulosa cells encoding the nuclear receptor transcription factor Pgr,which plays a key role in regulating the hypothalamic-pituitaryovarian axis in reproduction (Natraj & Richards, 1993).Several genes with differential RIs, includingtaf9,gtf2a1,ercc2, andtbpl2, were enriched in basic transcription factor pathways.In the testis, these genes all contained RI regions,thus providing insight into the characteristics of RIs among transcription factors in this organ.Tbpl2plays a key role in mouse oocyte development by regulating the transcription of oocyte-expressed genes (Gazdag et al., 2009; Yu et al.,2020).In fish,tbpl2is indispensable for embryonic development, but its role in germ cell development has not yet been elucidated (Bártfai et al., 2004).AS in mouse meiotic spermatocytes favors RIs (Naro et al., 2017).In our study,most differential RIs observed in the gonads originated in the testis, and some genes, such asstag2,stag3, andcdk7, were enriched in the cell cycle pathway.Bothstag2andstag3are components of the cohesion complex, which is necessary for sister chromatid cohesion (Nasmyth et al., 2000).STAG2 isrequired to repair DNA damage through homologous recombination in the S/G2 phase (Kong et al., 2014).Furthermore,stag3is a meiosis-specific gene expressed in the gonads and plays an important role in gametogenesis andfertility (Garcia-Cruz et al., 2010).The protein encoded bycdk7, which is a member of the cyclin-dependent protein kinase (CDK) family and the catalytic subunit of CDK-activated kinase (CAK), is also involved in RNA polymerase II-mediated RNA transcription (Shiekhattar et al., 1995).
RI genes were more up-regulated in testis compared to other AS types
A total of 61.32% (5 077/8 279) of the identified AS genes were differentially expressed in the gonads (Figure 4A), thus illustrating the close regulatory relationship between AS and gene expression.Our results showed that RI genes were more up-regulated (higher average expression) in the testis compared to other AS-type genes, which may be related to the process of meiosis in the testis.Recent studies on mouse testes have shown that meiotic intron-retaining transcripts(IRTs) are exclusively localized in the nucleus (possibly for later use) and show higher stability than other spliced transcripts (Naro et al., 2017).Thus, the up-regulation of RI genes in the testis of Chinese tongue sole may result from IRT storage.The effect of RIs on gene expression has also been illustrated by the down-regulation of non-physiologically relevant transcripts.For example, the steady-state expression levels of IRTs in mature neurons are significantly lower than that in murine embryonic stem cells during cell differentiation(Braunschweig et al., 2014).
AS may regulate differential expression of genes related to fish gonadal differentiation
Based on our results, 25 genes related to gonadaldifferentiation regulated by AS were screened, includingesrrbandwt1a.Theesrrbgene is known to encode estrogenrelated receptor b, which belongs to the NR3B subgroup(Tremblay & Giguère, 2007).Furthermore,esrrbis a transcription factor related to the self-renewal of embryonic stem (ES) cells, andesrrbknockout embryos die by embryonic day 10.5 (E10.5) (Luo et al., 1997).As an important regulatory gene,esrrbalso maintains the stemness of trophoblast stem cells (Latos et al., 2015).In addition,esrrbis involved in the proliferation of gonadal germ cells, withesrrbloss leading to a decrease in germ cell number (Mitsunaga et al., 2004).Our results showed thatesrrbtranscripts were highly expressed in the ovary of Chinese tongue sole and contained a female-specific exon (4th exon ofesrrb).The role ofesrrbin maintaining cell stemness suggests it may play a role in the development of ovarian germ cells.The Wilms’tumor suppressor genewt1encodes a zinc-finger transcription factor and plays an essential role in the development of the urogenital system in mice and humans (Morrison et al., 2006;Smolen et al., 2004).Twowt1paralogs,wt1aandwt1b, are found in fish.Bothwt1aandwt1bare important in zebrafish kidney development (Bollig et al., 2006).Furthermore,wt1aplays crucial roles in kidney development and sex determination in Nile tilapia, withwt1aknockdown in the kidneys resulting in developmental failure and non-expression of sex-determining genes in the gonad (Jiang et al., 2017).The retention of the 9th intron of thewt1atranscript in the Chinese tongue sole testis may promotewt1aexpression in this organ.
Regulation of gonadal differentiation by splicing regulators
Many splicing regulators in the gonads undergo sex-specific splicing.For example, inCaenorhabditiselegans, ~18% of splicing regulators are subject to AS during development(Barberan-Soler & Zahler, 2008).InDrosophilagonads,splicing regulators control cell type-specific splicing through sex- and stage-specific isoforms (Gan et al., 2010).Recent studies have shown that splicing regulators contribute to cell differentiation.Many splicing regulators are highly expressed in undifferentiated spermatogonia, indicating the active expression of splicing regulators and control of multiple processes by AS during cell differentiation (Liao et al., 2021).Splicing regulators, such assrsf1a,srsf1b, andu2af1, can modulate mRNA splicing.For example, the arginine/serinerich domain ofsrsf1can bind to U1 small nuclear ribonucleoprotein (snRNP) and help U1 snRNP to bind to the 5'-splice site containing pre-mRNA (Kohtz et al., 1994).In our study, twosrsf1paralogs were found in the gonads ofCynoglossus semilaevis, i.e.,srsf1aandsrsf1b, which showed different expression patterns.The DEAH/DEAD-box helicase family membersdhx35,dhx8, andddx5function as adenosine triphosphate (ATP)-dependent RNA helicases (Bourgeois et al., 2016).In addition to its involvement in the splicing regulation of pre-mRNA,ddx5plays a role in gonadal differentiation and is a novel androgen receptor-interacting protein (Clark et al., 2008).Furthermore,ddx5andddx17are master regulators of the estrogen and androgen signaling pathways and affect steroid hormone synthesis by regulating the transcription and splicing of genes up- and downstream of estrogen and androgen (Samaan et al., 2014).Recent research has also shown thatddx5is expressed in zebrafish gonads and mostddx5-deficient females develop into fertile males, with only a small proportion developing into infertile females, suggesting thatddx5is essential for oocyte maturation (Sone et al., 2020).In addition,ddx5also impacts male fertility, not only regulating the expression of cell cycle genes in spermatogonia but also regulating the proper splicing of genes required for spermatogenesis (Legrand et al., 2019).In the present study,ddx5was highly expressed in the testes,indicating thatddx5may also be involved in regulating testicular germ cell development and male fertility.We also found thattra2aandtra2bwere expressed at significant levels in the testes and may play important roles in spermatogenesis.TheDrosophila melanogasterhomolog,tra2, is essential for female sexual differentiation and male spermatogenesis (Amrein et al., 1988).In medaka (Oryzias latipes),tra2aandtra2bhave been detected in the germ cells of both sexes prior to sex differentiation, and may therefore be involved in this process (Shiraishi et al., 2004).Thetra2homolog (Mntra-2a) found in the oriental freshwater prawn(Macrobrachium nipponense) is highly expressed in the gonads of both sexes, mainly in oocytes and spermatocytes,and may play an important role in embryonic development and early gonadal development (Wang et al., 2019).
CONCLUSIONS
By sequencing the transcriptome ofC.semilaevis, we revealed extensive transcription and novel gene generation in the testis.SEs and RIs were the most common types of AS in fish gonads, consistent with findings in mammalian and bird gonads, suggesting that splicing is conservatively regulated across species.The DAS genes identified between the testis and ovary ofC.semilaeviswere primarily related to RNA splicing activity, indicating differential effects of sex-specific regulation of AS on gonadal differentiation.Notably, we observed differential RIs in mitosis- and meiosis-related genes in the testis, suggesting that AS may also participate in the regulation of testicular germ cell development.Moreover, we identified several sex-specific isoforms related to gonad and germ cell development, which will facilitate research on the functions of spliced isoforms.In conclusion, AS in the gonads participates in sexual development by regulating splicing of sex-related genes and splicing regulators implicated in gonadal differentiation.Coordinated regulation of AS and splicing regulators contributes significantly to gonadal differentiation.
DATA AVAILABILITY
The raw RNA-Seq reads are available in the NCBI SRA(BioProjectID PRJNA778900), GSA (accession No.CRA006156), and Science Data Bank databases (data doi:10.11922/sciencedb.01543).The Iso-Seq reads are available in the SRA (BioProjectID PRJNA778651) and Science Data Bank databases (data doi: 10.11922/sciencedb.01543).
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
Q.W.and C.W.S.designed the research.Y.F.L.and K.Q.L.analyzed the data and wrote the manuscript.Q.L.and H.Y.W.performed histological sectioning and validation experiments.Y.F.L., Q.W., K.Q.L., H.Y.W., C.H.L., and C.W.S.revised the manuscript.All authors read and approved the final version of the manuscript.
杂志排行

Zoological Research的其它文章
- Mechanism of hyperproteinemia-induced blood cell homeostasis imbalance in an animal model
- Phylogenetic relationships of the zokor genus Eospalax(Mammalia, Rodentia, Spalacidae) inferred from wholegenome analyses, with description of a new species endemic to Hengduan Mountains
- MonkeyTrail: A scalable video-based method for tracking macaque movement trajectory in daily living cages
- Unknown species from China: The case of phrurolithid spiders (Araneae, Phrurolithidae)
- A new species of the gudgeon genus Microphysogobio Mori, 1934 (Cypriniformes: Cyprinidae) from Zhejiang Province, China
- High-quality chromosome-level genome assembly of Tibetan fox (Vulpes ferrilata)