荧光分光光度法测定水中石油类方法研究
2022-06-06郭晶晶关玉春
郭晶晶,赵 莉,关玉春,王 琳
(天津市生态环境监测中心,天津 300191)
石油类主要是由烃类化合物组成的一种复杂混合物[1],在《国家危险废物名录》中列第8 位[2]。石油类物质对水的色、味和溶解氧有较大影响,其致癌、致畸、致突变物质可通过食物链传递给人体,危害人体健康[3-5],是水污染物排放标准中常见的污染物控制项目。目前,石油类测定方法主要有荧光分光光度法、红外法、紫外法、重量法和气相色谱法。红外法所用的萃取剂四氯乙烯不稳定,提纯过程是公认的技术难点;紫外法抗干扰能力欠佳;重量法检出限高,操作繁琐,仅适用于重污染水体的监测;气相色谱法通常用于某个组分的定性分析,且仪器造价高、不具有普适性[6-12]。
荧光分光光度法灵敏度高,选择性好,测定过程快速准确[13]。但现行的相关标准及检测方法一般选用二氯甲烷、石油醚或环已烷作为萃取剂。二氯甲烷被世界卫生组织列入致癌物清单;石油醚是低级烷烃的混合物,属易燃易爆物质;环己烷属于极易燃物质,具有强烈刺激性气味,危害大。《水质 石油类的测定—分子荧光光度法》(SL 366—2006)尽管选用了正己烷作为萃取剂,但萃取液用量大,过程复杂,缺少硅酸镁吸附分离极性物质的步骤,不适用于测定基体复杂的样品。本研究结合石油类分析技术发展趋势和国内监测机构能力现状,通过优化实验条件,建立荧光分光光度法测定水中石油类的方法,能适应我国绝大部分环境监测及相关实验室的仪器设备、技术能力,满足各项管理需求。
1 实验部分
1.1 试剂和仪器
试剂:正己烷(色谱纯,迪马科技有限公司)、无水硫酸钠(于550 ℃下灼烧4 h,冷却备用,天津光复科技发展有限公司)、硅酸镁(于550 ℃下灼烧4 h,冷却后按6%(m/m)的比例加入蒸馏水,密塞并充分振荡数分钟,放置12 h,备用,天津科密欧化学试剂有限公司)、石油类标准贮备液(市售正己烷体系)。
仪器:RF5301 型荧光分光光度计(日本岛津公司)、比色皿(10 mm 石英荧光比色皿)、分液漏斗(1000 mL,聚四氟乙烯旋塞)、锥形瓶(50 mL,具塞磨口)、水平振荡器、离心机(配备玻璃离心管)。
1.2 实验原理
用正己烷萃取样品中的油类物质,经无水硫酸钠脱水后,再用硅酸镁吸附除去动植物油类等极性物质,其中的石油类物质经激发光源照射,分子产生跃迁,当分子从激发态返回到基态的振动能级时,以荧光形式释放吸收的能量发出分子荧光。荧光强度在一定浓度范围内与石油类含量成正比。
1.3 实验步骤
(1)将水样转移至分液漏斗中,用正己烷充分振荡萃取,静置分层后,将下层水相全部转移至量筒中,测量样品体积并记录,乳化程度较重时,需进行破乳处理。
(2)将上层萃取液转移至已加入无水硫酸钠的锥形瓶中,盖紧瓶塞,振荡数次,静置。若无水硫酸钠全部结块,需补加无水硫酸钠直至不再结块。也可将萃取液通过已放置好无水硫酸钠的玻璃漏斗脱水。
(3)继续向萃取液中加入硅酸镁,置于振荡器上振荡,静置沉淀。在玻璃漏斗底部垫少量玻璃棉,过滤,取滤液进行比色定量。
通过测定自配样品、标准样品和实际样品,确定最佳条件参数,包括萃取剂(正己烷)用量与水样体积的比例、萃取时间、萃取次数、破乳方式、脱水方式、极性物质的吸附分离方式、测定波长等。
2 结果与讨论
2.1 标准曲线的建立
标准物质采用HJ 油标准(编号GBW(E)080913,成分是20 号重柴油和润滑油),用正己烷稀释。在激发波长为310 nm、发射波长为360 nm的条件下,使用1 cm 比色皿,以正己烷作参比,测定荧光强度。以石油类浓度(mg/L)为横坐标,相应的荧光强度为纵坐标,建立标准曲线,见表1。由表1 可知,标准曲线的截距a=-1.76,斜率b=29.8,相关系数r=0.9999。利用标准样品(批号BW012,保证值为5.00 mg/L,不确定度为±7%mg/L)对标准曲线进行验证,测定结果为4.95mg/L,表明标准曲线的准确性良好。

表1 标准曲线Tab.1 Standard curve
2.2 萃取剂(正己烷)与水样体积的比例
用萃取剂定量提取水中的石油类物质,涉及到分配系数问题。《水质 石油类的测定 分子荧光光度法》(SL 366—2006)中给出的萃取剂与水的体积比为1:10,《水和废水监测分析方法(第3版)》中给出的体积比为1:10,《海洋监测规范 第4 部分:海水分析》(GB 17378.4—2007)中给出的体积比为1:25,《生活饮用水标准检验方法》(GB/T 5750.7—2006)中给出的体积比为1:10,文献中给出的体积比也多为1:10、1:20、1:40。
为了使萃取剂正己烷用量既能达到定量萃取的要求,又能节省试剂,进行如下实验:选择不同体积的正己烷、配制样品和实际样品,添加不同浓度的石油类标准溶液进行加标回收率实验,结果见表2 和表3。
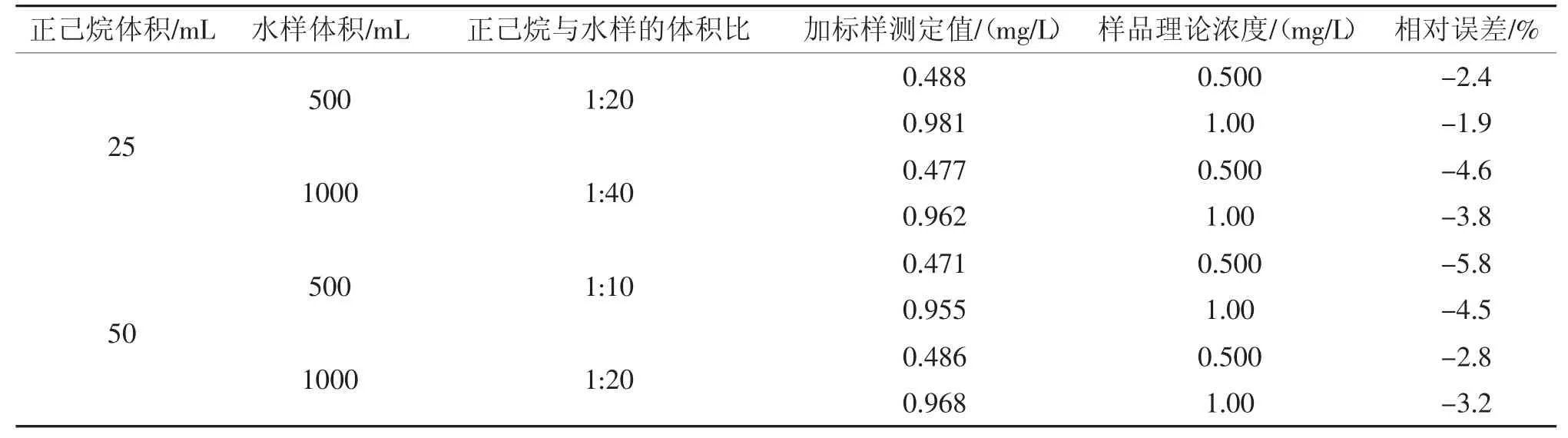
表2 萃取剂(正己烷)与水样的体积比对石油类测定的影响(配制样品)Tab.2 Influence of ratio of extractant(n-hexane) and water sample volume on determination of petroleum(prepared sample)
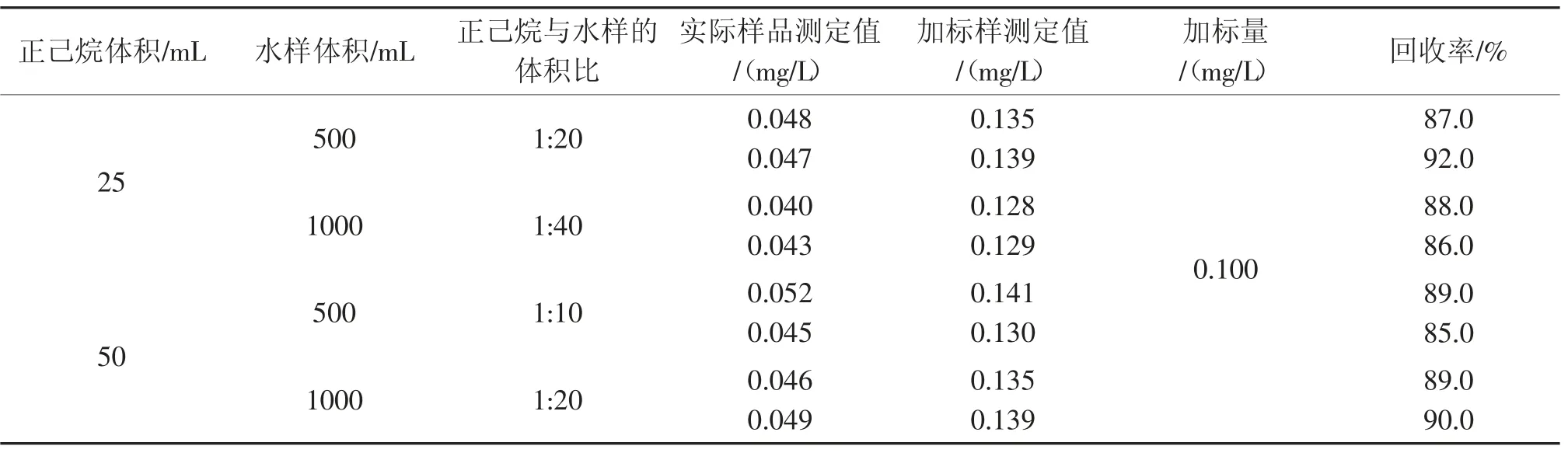
表3 萃取剂(正己烷)与水样的体积比对石油类测定的影响(实际样品)Tab.3 Influence of ratio of extractant(n-hexane) and water sample volume on determination of petroleum(practical example)
由表2 和表3 可以看出,萃取剂(正己烷)与水样的体积比为1:40 时,配制样品测定的相对误差为-4.6%~-3.8%,实际样品的加标回收率为86.0%~88.0%;体积比为1:20 时,配制样品测定的相对误差为-3.2%~-1.9%,实际样品的加标回收率为87.0%~92.0%;体积比为1:10 时,配制样品测定的相对误差为-5.8%~-4.5%,实际样品的加标回收率为85.0%~89.0%。提高萃取剂(正己烷)与水样的体积比,加标回收率没有明显提高。因此,从降低检出限、节约试剂和可操作性的角度考虑,选择水样体积为500 mL,萃取剂(正己烷)的体积为25 mL。
2.3 萃取时间
石油类测定的关键环节是萃取过程,萃取振摇时间直接影响萃取效果,选择振摇时间为0.5 min、1 min、2 min、3 min、5 min 进行实验,通过实际样品加标实验以检验最佳萃取时间,结果见表4 和图1。
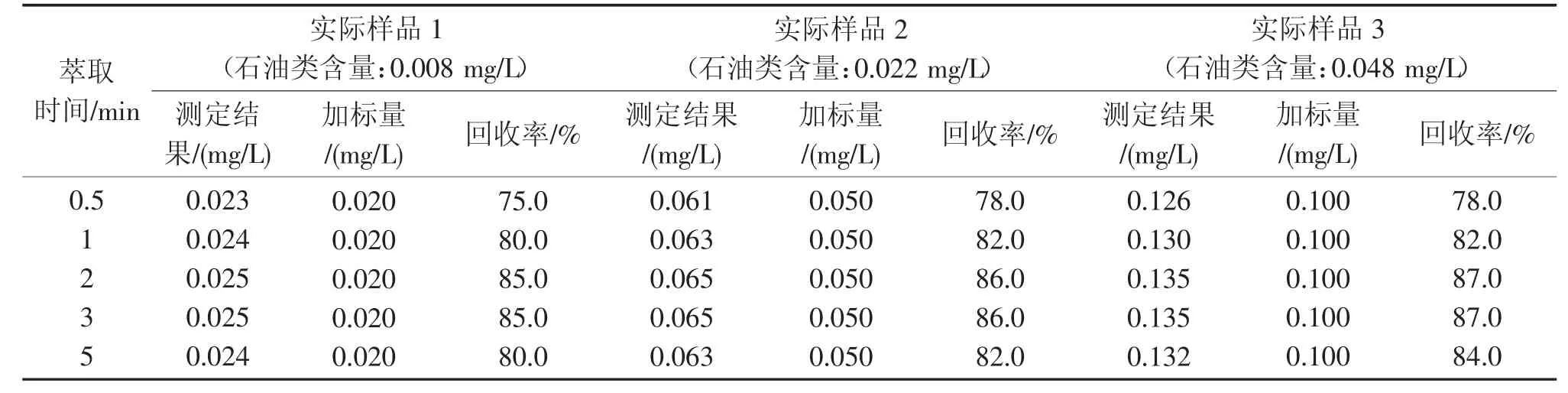
表4 不同萃取时间对实际样品加标回收率的影响Tab.4 Effect of different extraction time on practical example recovery
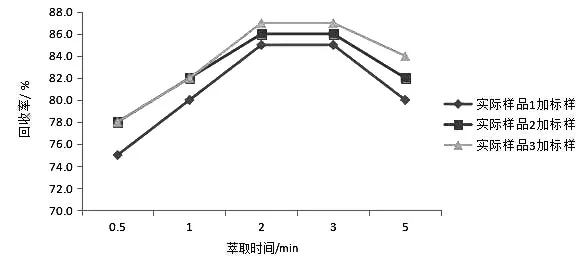
图1 不同萃取时间的实际样品加标回收率Fig.1 The practical example recovery rate of sample with different extraction time
由表4 和图1 可见,萃取时间小于2 min 时,实际样品的加标回收率较低,2 min 时达到最高;3 min 时变化不大,基本与2 min 时的结果相同;5 min时明显下降。这是由于随着萃取振摇时间的延长,样品乳化程度增加,易造成回收率降低。因此将萃取时间确定为2 min。
2.4 萃取次数
为了确定萃取次数与回收率之间的关系,开展以下萃取试验。
(1)1 次萃取:分别取3 个500 mL 实际水样于3 个1000 mL 分液漏斗中,分别加入不同浓度的石油类标准溶液(加标量为样品本底值的0.5~3 倍),再加入25.0 mL 正己烷,充分振荡2 min,重复测定6 次。
(2)2 次萃取:分别取3 个500 mL 实际水样于3 个1000 mL 分液漏斗中,分别加入不同浓度的石油类标准溶液(加标量为样品本底值的0.5~3 倍)。先加入15.0 mL 正己烷,再加入10.0 mL 正己烷,重复测定6 次。
比较1 次萃取和2 次萃取的加标回收率差异,结果见表5 和表6。
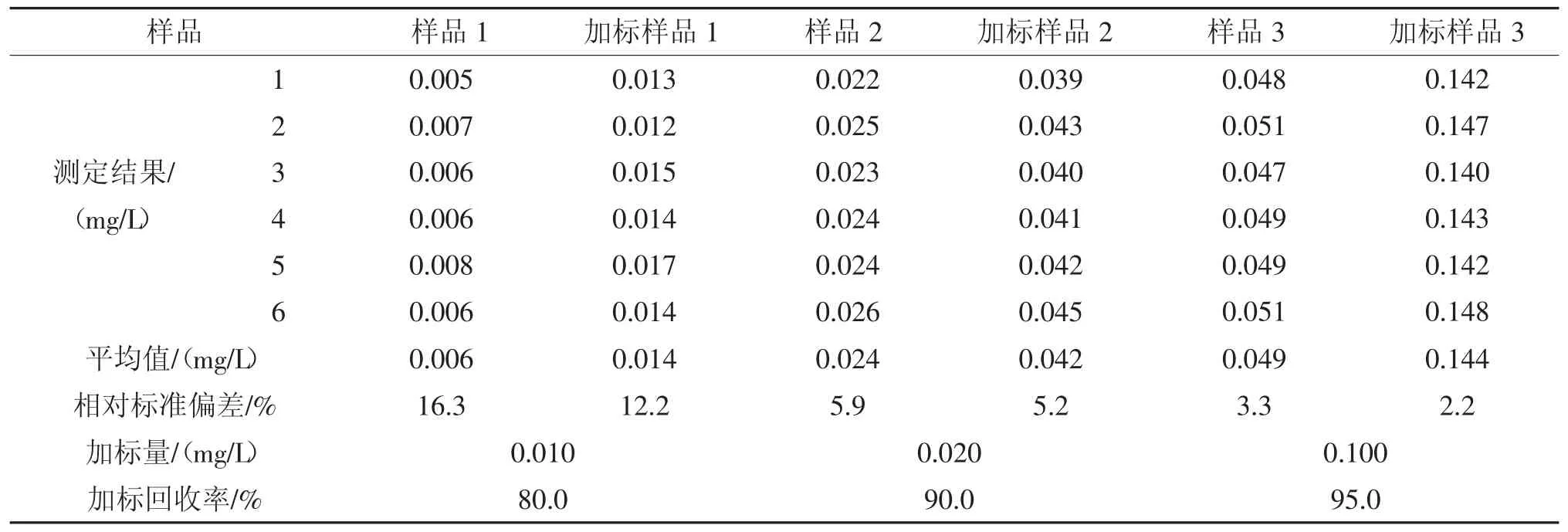
表5 1 次萃取实际样品回收率测定结果Tab.5 Determination results of practical example recovery rate after one extraction
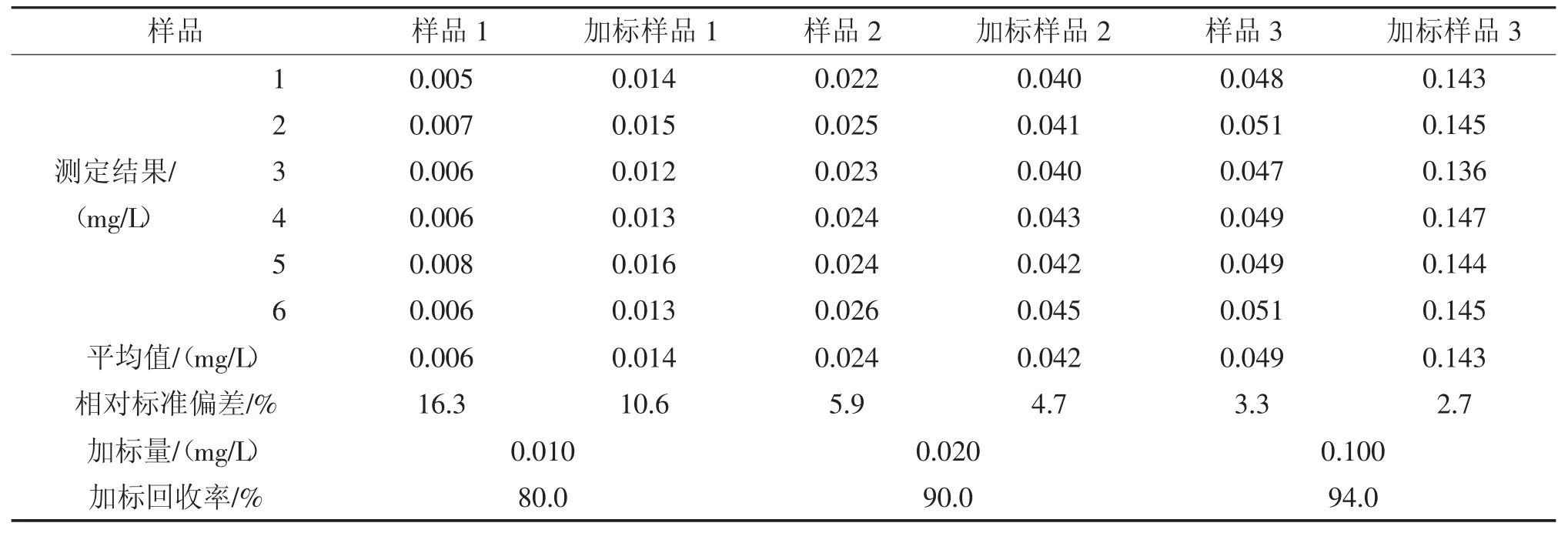
表6 2 次萃取实际样品回收率测定结果Tab.6 Determination results of practical example recovery rate after two extraction
由表5、表6 可见,1 次萃取的回收率为80.0%~95.0%,2 次萃取的回收率为80.0%~94.0%,二者没有明显差异,均能满足需要。这是因为当被萃取物质在水和萃取剂两相中的分配系数小于0.01 或大于100 时,萃取1 次,萃取效率已基本达到最大值。因此,为简化步骤,提高效率,采取1 次萃取方式。
2.5 自动萃取实验
目前大部分实验室均配备了自动萃取装置,既能节省人力,又能提高工作效率。采用垂直振荡器,通过配制样品检验自动萃取的效果,萃取速度为180~200 r/min,其他条件同手动萃取,结果见表7。
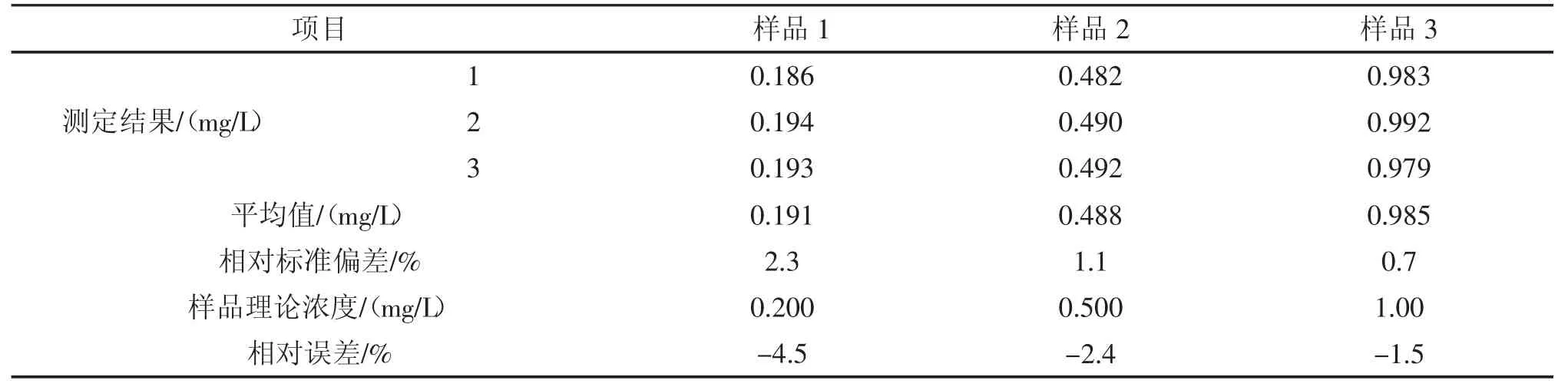
表7 自动萃取对配制样品测定结果的影响Tab.7 Influence of automatic extraction on the determination of prepared samples
由表7 可见,自动萃取样品测定结果的相对标准偏差为0.7%~2.3%,相对误差为-4.5%~-1.5%,能够满足实验要求。
2.6 破乳方式
破乳是油类萃取过程经常面临的难题,对于地下水或者清洁的地表水,萃取过程乳化现象较轻,但是对于浑浊的地表水或者污水,乳化现象较为严重,影响分层。破乳的方法主要有:(1)静置;(2)机械破乳;(3)加入电解质;(4)酸化;(5)加乙醇;(6)离心;(7)超声等。
针对某乳化现象较轻和较重的2 个水样,分别采用方法(1)、(2)进行破乳,效果不明显。采用方法(3)、(4)、(5),分别向500 mL 水样中加入10 g氯化钠、5 mL 浓硫酸、5 ml 浓盐酸和5 mL 无水乙醇,再加入25 mL 正己烷萃取,发现氯化钠对乳化较轻的水样效果明显,其余处理乳化层仍大量存在。直接向萃取液中加入上述4 种物质,发现前3个处理仍无效果,但乙醇破乳效果显著,且乙醇在荧光分光光度计的激发波长为310 nm、发射波长为360 nm 处无响应,因此不会干扰测定结果,一般加入1~4 滴即能达到理想的破乳效果。
考察离心的破乳效果时,将3 个水样(1 个较清洁水样和2 个污染较重的水样)的萃取液转入玻璃离心管中,2000 r/min 离心3min,乳化层即明显分离,破乳率随离心转数的增加而增大,也随作用时间的延长而增大。
考察超声的破乳效果。实验发现,超声处理后,从液滴的凝聚、沉降到分层必须经过较长的时间,且必须严格控制温度,操作较为繁琐,超声的破乳效果也不如离心明显,因此不建议采用。
综上所述,在样品乳化程度较重时可选择加入1~4 滴乙醇的方式破乳;若效果仍不理想,可将萃取液转移至玻璃离心管中,转速为2000 r/min,离心3 min。
2.7 萃取液脱水方式
萃取液脱水方式主要有2 种:(1)将萃取液经过“放置约10 mm 厚度无水硫酸钠的玻璃砂芯漏斗”进行脱水;(2)直接向萃取液中加入无水硫酸钠进行脱水。
方式(1)用10 mm 厚度无水硫酸钠的玻璃砂芯漏斗过滤,速度较慢,特别是表面无水硫酸钠吸水结晶后,发生堵塞,过滤不能正常进行,长时间的过滤造成萃取液中低沸点组分挥发损失,尽管正己烷属于低毒性物质,但长时间的过滤,对分析人员的健康会产生一定的危害。
方式(2)对于发生轻度乳化的样品,破乳和干燥可以同时完成,此脱水过程是在封闭的体系中完成的,减少了溶剂的挥发,减轻了对分析人员的健康危害,操作性更强。
分别取2 种不同水样500 mL 于1 000 mL分液漏斗中,分别加入不同浓度的石油类标准溶液(加标量为为样品本底值的0.5~3 倍),按照两种脱水方法对石油类进行测定,重复测定6 次。计算加标回收率,结果见表8。
由表8 可知,对于清洁水样,萃取时样品乳化程度较轻,脱水方式(1)、方式(2)的加标回收率都是80.0%;对于污染较重的水样,乳化程度较高,脱水方式(1)、方式(2)的加标回收率分别为84.0%和94.0%,方式(2)明显优于方式(1)。因此,建议采用方式(2)对萃取液进行脱水,减少溶剂挥发,减轻对分析人员的健康危害,可操作性也更强。但对于清洁水样,也可根据操作习惯,选择方式(1)进行脱水。
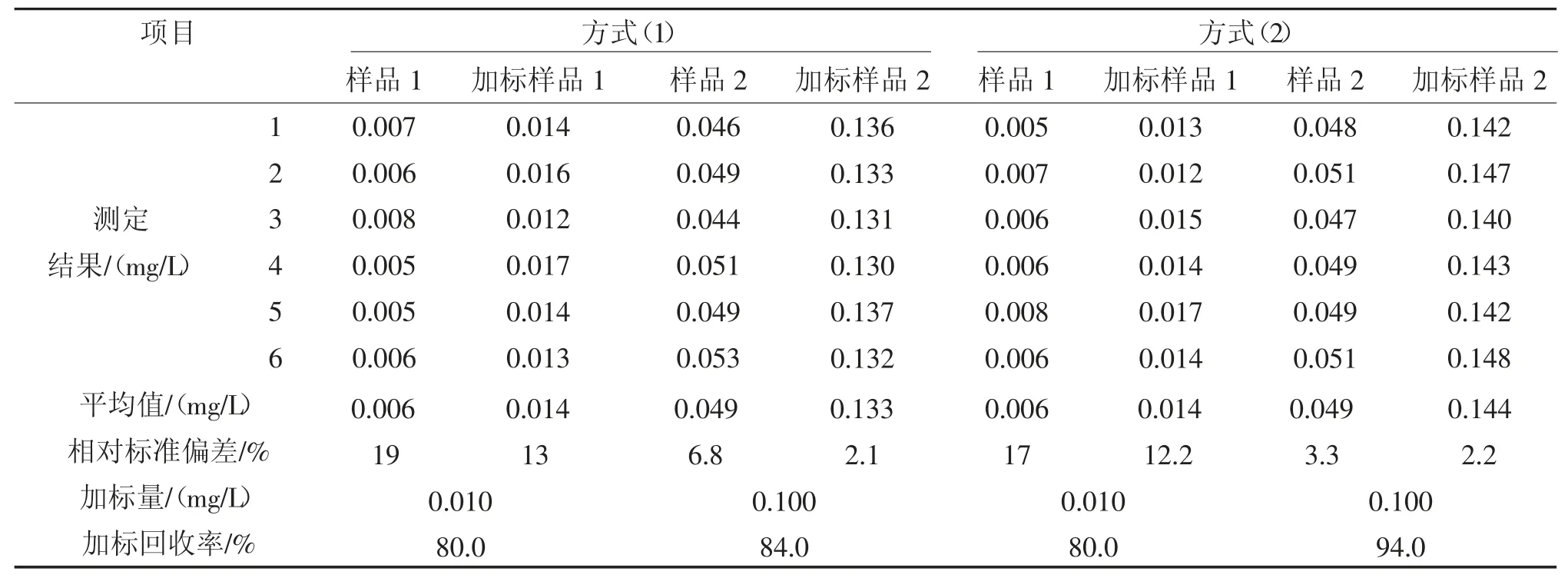
表8 2 种脱水方式对样品加标回收率的影响Tab.8 Effects of two dehydration methods on the standard recovery of samples
2.8 动植物油等极性物质的吸附分离方法
荧光分光光度法测定的原理是经紫外光激发,测定油中可发荧光的芳烃部分,对饱和烃类基本无响应。动物油主要含饱和脂肪酸,而植物油主要含不饱和脂肪酸,均无芳烃组分,理论上无荧光响应。将几种常见动植物油类(猪油、调和油、花生油、硬脂酸)取适量溶于正己烷,在激发波长为310 nm、发射波长为360 nm 条件下测定荧光强度,结果见表9。

表9 4 种动植物油类的荧光法测定结果Tab.9 Fluorimetric determination of 4 kinds of animal and plant oils
由表9 可知,对于理论浓度为100 mg/L 的样品,4 类动植物油类的荧光分光光度法测定结果均低于检出限。红外法测定结果的相对偏差为4.3%~21%,与样品理论浓度较接近。
由以上分析可知,荧光分光光度法不适于动植物油类的测定。但是某些非动植物油的极性物质(如含-C=O、-OH 基团的极性化学物质)也会产生荧光响应,从而导致测定结果偏高。
纵观各标准和文献,均以硅酸镁为吸附剂,吸附去除萃取液中动植物油类等极性物质。吸附分离方式主要有两种:
(1)吸附柱分离法:层析柱内径为10 mm、长度为200 mm、硅酸镁填充高度为80 mm、硅酸镁重量约为15.6 g,将脱水后的萃取液经过此柱吸附分离。
(2)振荡吸附分离法:向萃取液中直接加硅酸镁,然后通过摇床振荡吸附,振荡后的溶液静置沉淀,上清液经玻璃砂芯过滤后比色。
方法(1)中的吸附柱分离速度为0.5 mL/min~1 mL/min,通常分离20 mL 萃取液需要30 min 左右,正己烷是易挥发溶剂,不宜在开放的体系中进行吸附分离。
方法(2)中的摇床振荡容易实现批量分离,操作简单,分离过程在封闭的体系中进行,避免溶剂挥发,可操作性更高。此外,连续振荡使极性物质与硅酸镁充分接触,且振荡的力度和幅度较大,因此吸附效果更好。
用HJ 油标准物质、调和油配制成不同浓度的25 mL 正己烷溶液,不经萃取、脱水等过程,直接用2 种吸附方式分离极性物质后进行测定,测定结果见表10。
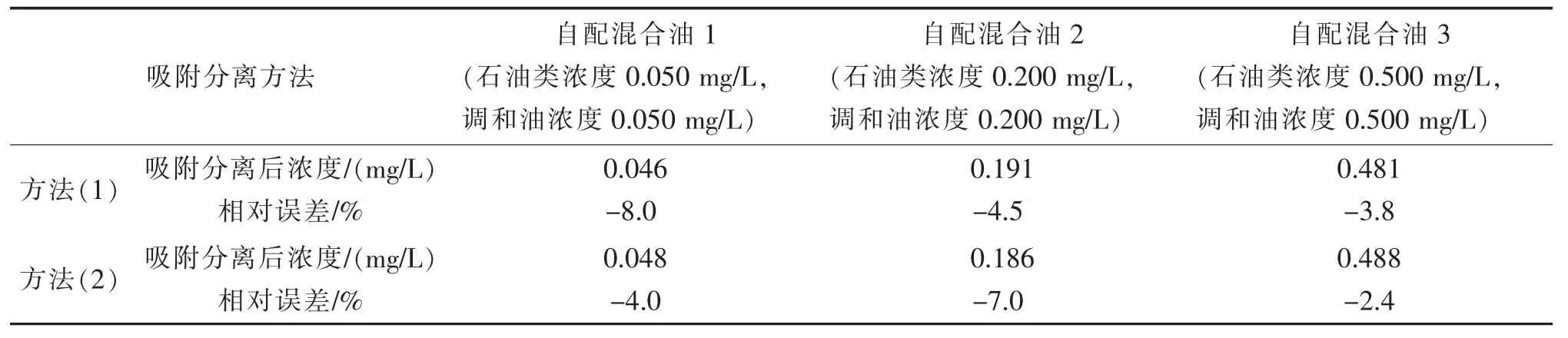
表10 两种吸附分离方式对样品测定结果的影响Tab.10 The influence of two adsorption separation methods on the determination results of samples
由表10 可见,方法(1)的相对误差为-8.0%~-3.8%,方法(2)的相对误差为-7.0%~-2.4%,没有显著差别。操作者可以根据实验室的实际情况选择方法(1)或方法(2)来进行极性物质的吸附分离。
2.9 测定波长的确定
荧光分光光度法测定的理论基础是某些物质在特定波长激发光和发射光下会发出荧光,例如,腐殖质在激发波长为230~260 nm 范围内会发出320~350 nm 范围的荧光,蛋白质在220 nm和275 nm 激发光下会发出300~350 nm 范围的荧光。此外,多环芳烃在波长为245~280 nm 激发光照射下,发出波长为310~400 nm 的荧光,荧光波长决定于芳烃的环数。
石油类的测定也基于此,石油的主要组成部分中约有95%~99%的成分是由碳氢元素组成的烃类,分为环烷烃、烷烃、芳香烃3 类物质,分别属于脂环族、脂肪族和芳香族,其结构复杂且稳定。此外石油中还含有非烃化合物,例如含硫化合物,以环烷酸形式存在的含氧化合物以及存在于高沸点馏分和渣油中的胶状、沥青状物质。其有荧光响应的是其中的芳烃部分,而饱和烃类基本无响应。地表水(湖库水、景观水、排污河)、钢铁行业废水、化工行业废水样品的荧光光谱图见图2~图6。
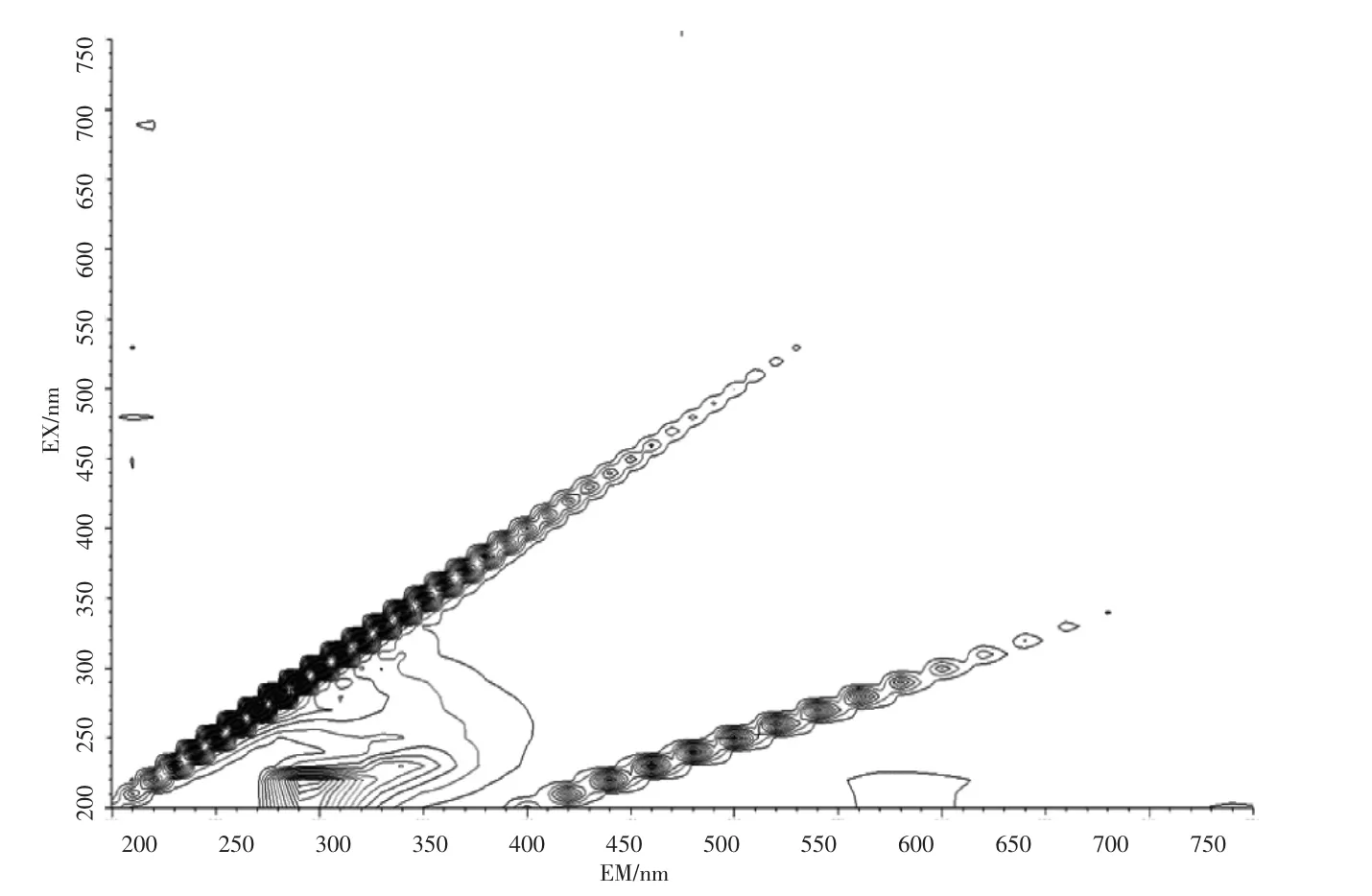
图2 湖库水扫描图Fig.2 Lake reservoir water scan
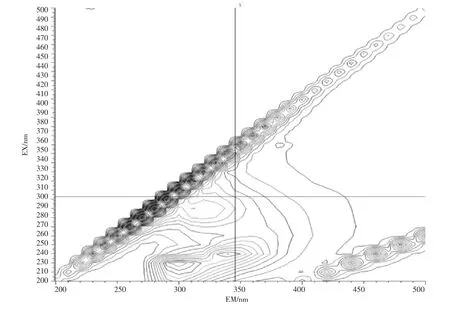
图3 排污河扫描图Fig.3 Scan of drainage rive
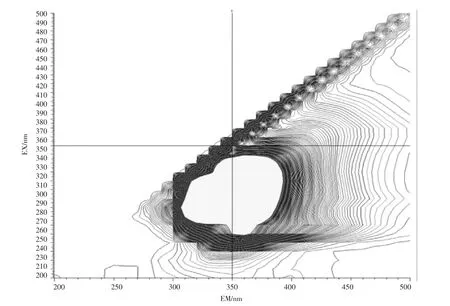
图4 景观水扫描图Fig.4 Landscape water scan
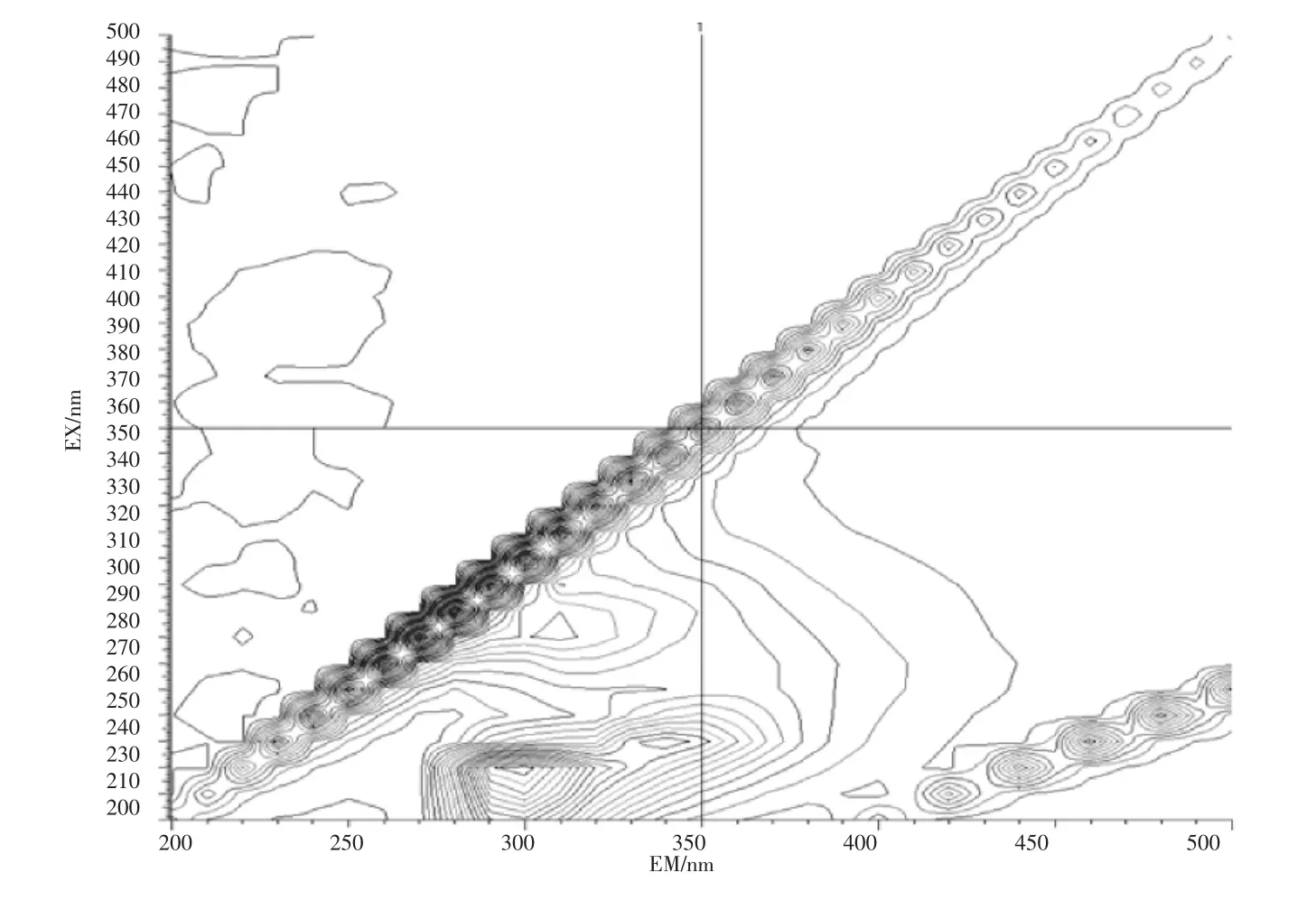
图5 钢铁行业废水扫描图Fig.5 Iron and steel industry waste water scan
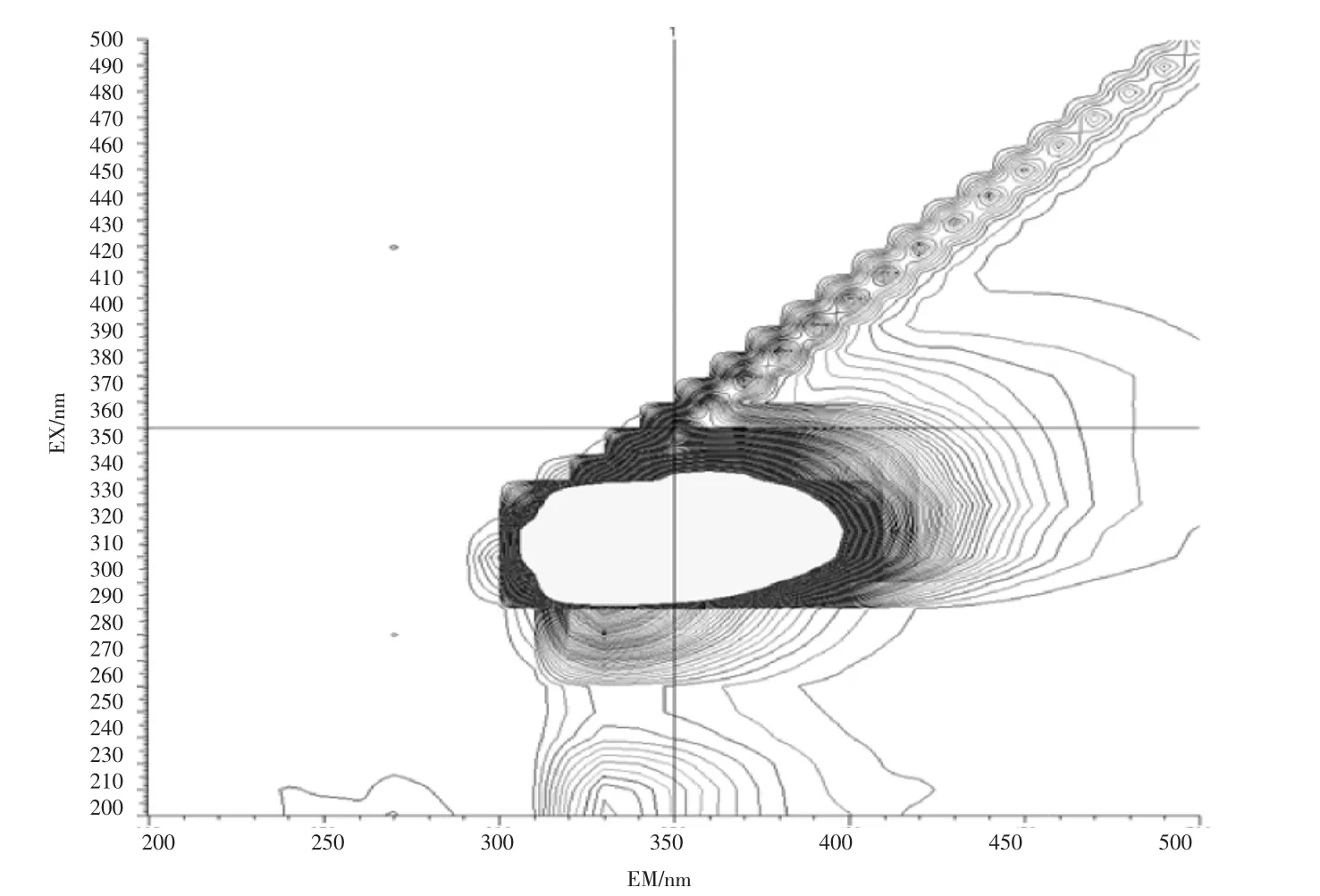
图6 化工行业废水扫描图Fig.6 Scan of chemical industry wastewater
由图2~图6 的扫描结果可知,由于水质样品种类繁多,基体差异很大,最大吸收峰位置也各不相同,但都集中在300~400 nm 之间。标准油是以正己烷为溶剂、20 号柴油为主要成分的市售标准物质或标准样品,标准样品的作业指导书明确要求测定波长为:激发波长为310 nm、发射波长为360 nm。为了保持方法的统一性和延续性,拟选择测定波长为:激发波长为310 nm、发射波长为360 nm。
2.10 方法检出限的确定
按照《环境监测 分析方法标准制订技术导则》(HJ 168—2020)中附录A.1.1 方法确定石油类测定的检出限。即重复测定空白样品7 次,并计算7 次平行测定的标准偏差,按公式(1)计算方法检出限,以4 倍检出限作为方法测定下限,结果见表11。
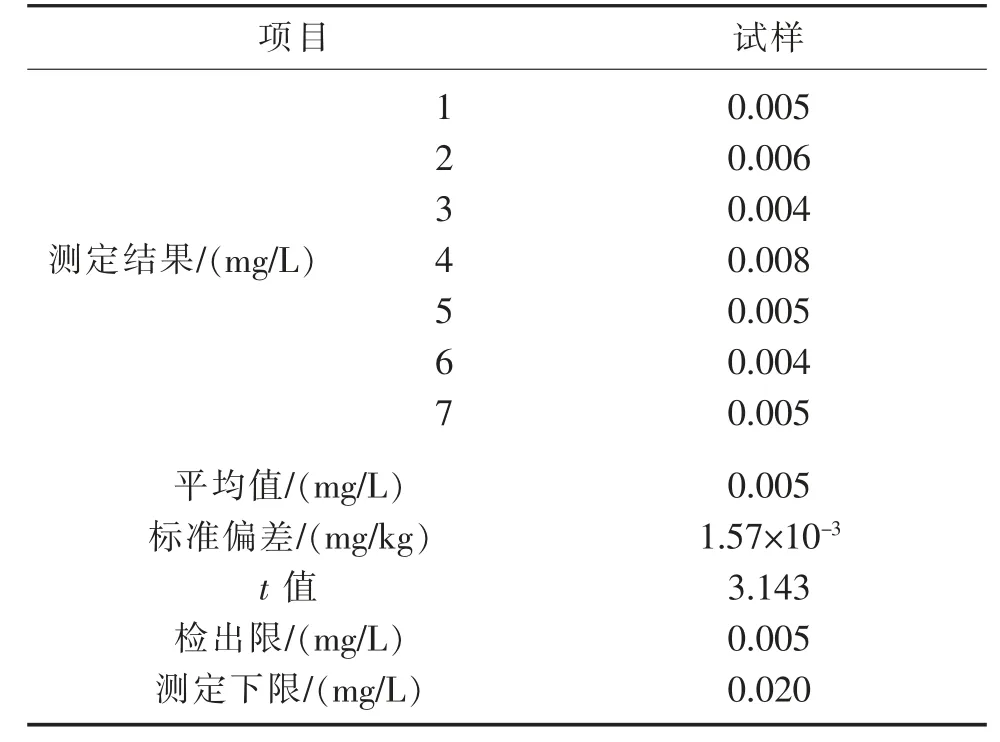
表11 方法检出限、测定下限测试数据表Tab.11 Methods test data table of limit of detection and limit of determination

式(1)中:MDL 为方法检出限;n 为样品的平行测定次数;t 为自由度为n-1、置信度为99%时的t 分布(单侧);s 为n 次平行测定的标准偏差。
由表11 可见,当取样体积为500 mL,萃取剂(正己烷)体积为25 mL,使用10 mm 比色皿时,方法检出限为0.005 mg/L,测定下限为0.020 mg/L。该检出限能满足地表水Ⅰ、Ⅱ、Ⅲ类的水质监测要求(地表水Ⅰ、Ⅱ、Ⅲ类石油类限值为0.05 mg/L),也能满足海水第一、二类的水质监测要求(限值为0.05 mg/L)。
2.11 方法精密度
向500 mL 空白水样中分别加入0.25 mL、0.50 mL、2.00 mL、5.00 mL 石油类标准使用液(100 mg/L),配制成浓度为0.050 mg/L、0.100 mg/L、0.400 mg/L、1.00 mg/L 共4 个样品,按样品分析步骤全程序,每个浓度样品平行测定6 次。计算每个浓度的平均值、相对标准偏差、加标回收率,结果见表12。
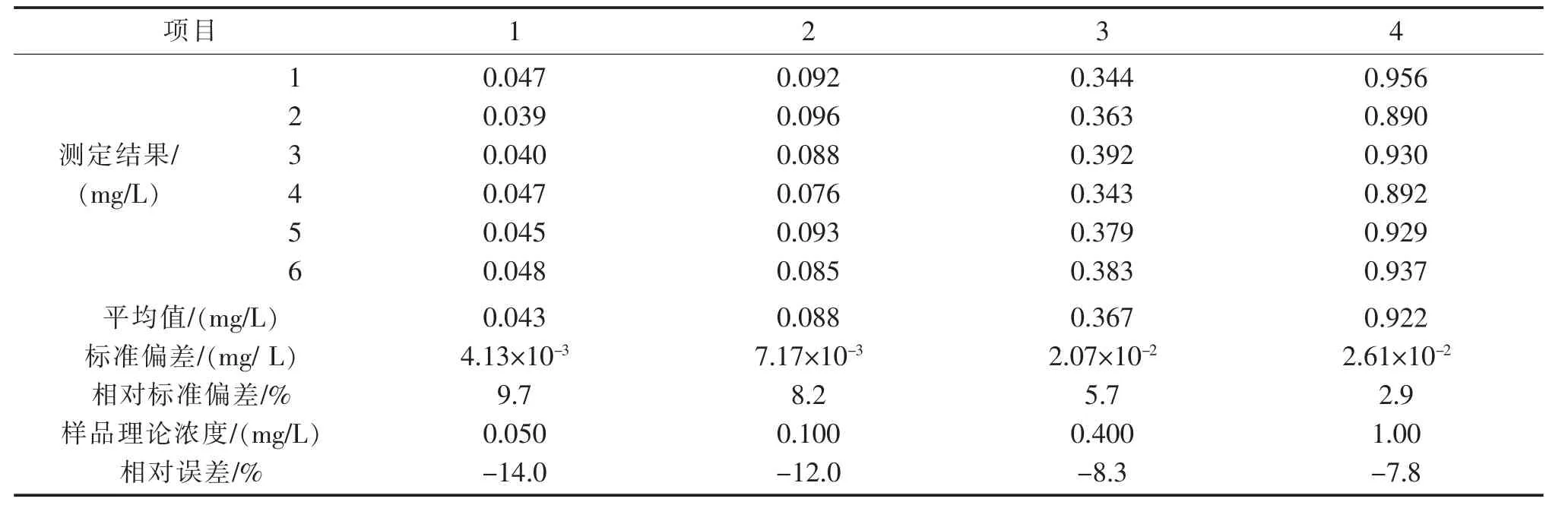
表12 配制样品精密度测定结果Tab.12 Test results of precision of prepared samples
通过重复测定有证标准样品,计算相对标准偏差的方式考察方法的精密度,测定结果见表13。
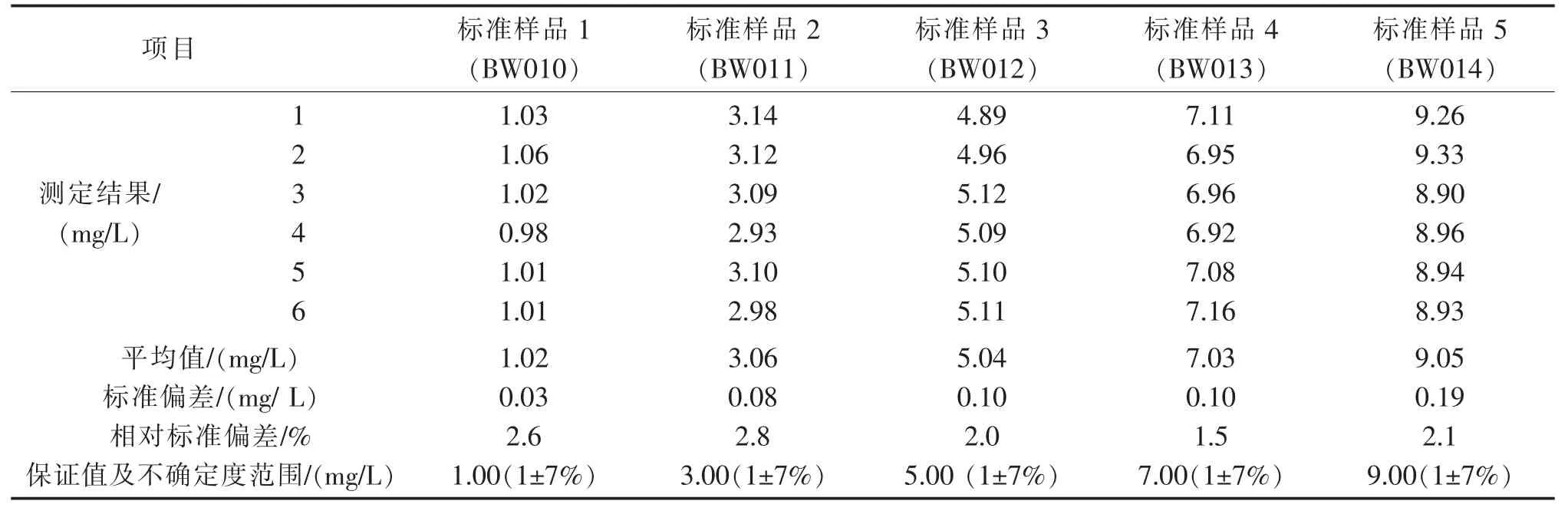
表13 有证标准样品测定结果Tab.13 Test results of standard samples
由表12 可见,4 个配制样品测定的相对标准偏差为2.9%~9.7%,相对误差为-14.0%~-7.8%。由表13 可以看出,有证标准样品6 次重复测定的相对标准偏差在1.5%~2.8%之间,精密度良好。
2.12 方法正确度
向实际样品(地表水和地下水)中分别加入不同体积的石油类标准使用液(100 mg/L)(实际样品加标量为其测定值的0.5~3 倍)进行加标回收率实验,按样品分析步骤全程序,每个样品平行测定6 次。计算平均值、相对标准偏差、加标回收率,结果见表14。
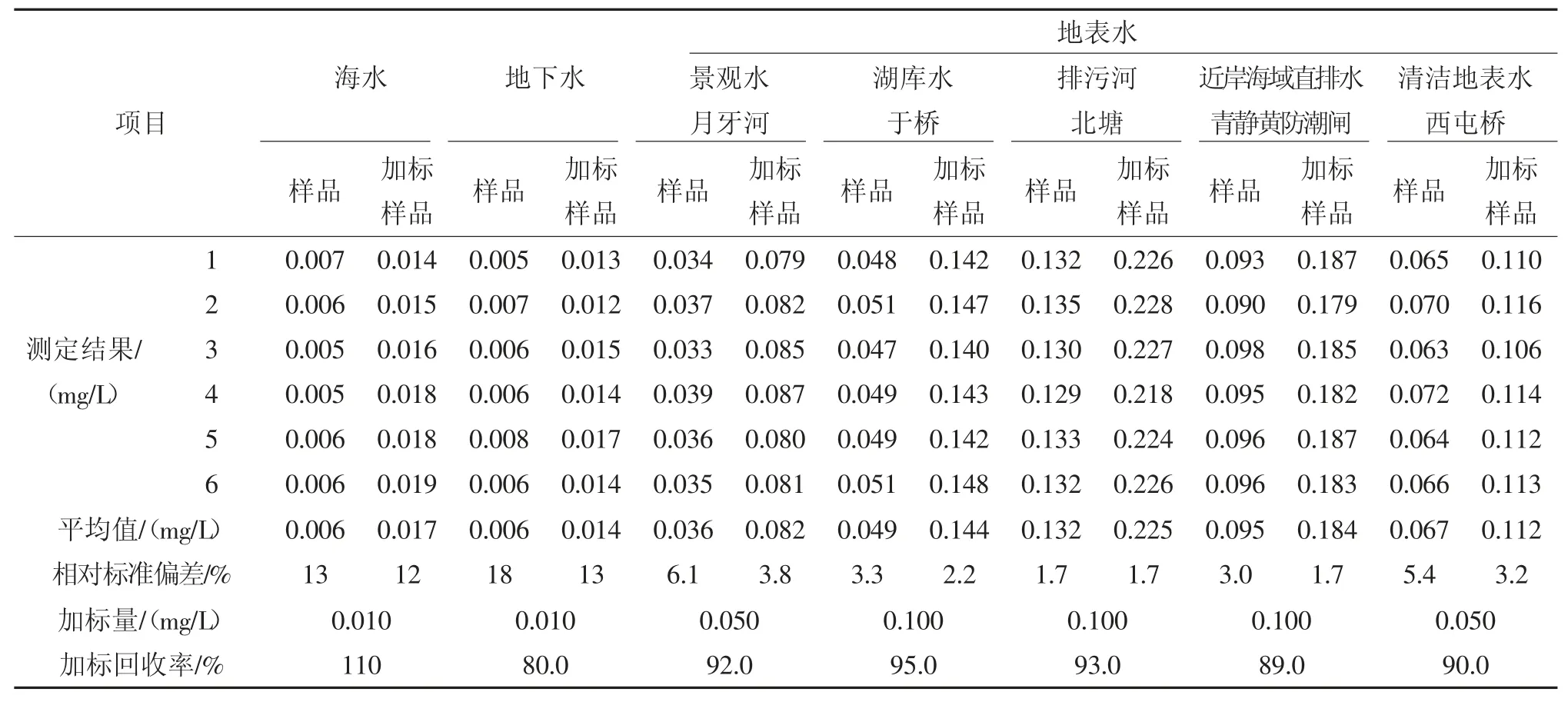
表14 实际样品加标测定结果Tab.14 Determination results of the actual sample recovery rate
由上表可知,实际样品的加标回收率在80.0%~110%之间。由表13 可以看出,有证标准样品的6 次重复测定结果均在给定的保证值范围内,方法正确度良好。
3 结论
建立了荧光分光光度法测定石油类的方法,适用范围包括地表水、地下水和海水。最佳实验条件是:当水样体积为500 mL 时,萃取剂(正己烷)体积为25 mL,振摇萃取2 min,萃取1 次,滴加无水乙醇或离心的方式破乳,直接向萃取液中加入无水硫酸钠进行脱水,加入硅酸镁吸附分离极性物质,选择激发波长为310 nm、发射波长为360 nm,10 mm 光程的比色皿进行分析测试。此方法检出限为0.005 mg/L,相对标准偏差范围为1.5%~9.7%,加标回收率为80.0%~110%。满足国内相关环境质量的要求,是一种易于推广使用的水质石油类分析方法。
