先天性巨大黄斑缺损1 例△
2022-05-29姚帮桃刘刚陈旭剑赵孝贵
姚帮桃 刘刚 陈旭剑 赵孝贵
(1.南京市溧水区人民医院 东南大学附属中大医院溧水分院眼科 南京 211200;2.南京市溧水区中医院眼科 南京 211200)
资料患者女性,40岁,双眼自儿童期视物模糊至今。否认双眼外伤及手术史,否认有全身系统性疾病。父母非近亲结婚,其母亲孕期感染史不详。眼科检查:双眼最佳矫正视力(best corrected visual acuity, BCVA) +0.50:0.15,双眼前节未见明显异常,眼压正常。
眼底示双侧视盘色淡红,边界清,右、左眼黄斑区各可见约3×4视盘直径大小的边界清楚的椭圆形和圆形视网膜脉络膜凹陷病灶,其边缘及底部见较多色素沉着,底部见裸露的脉络膜大血管,视网膜血管在其边缘走行正常(图1)。红外眼底(infrared fundal, IR)可见双眼黄斑区清晰病灶轮廓(图2)。
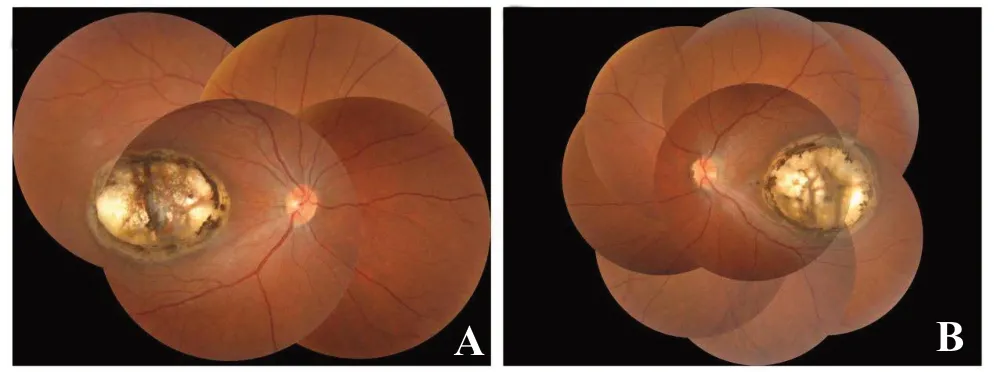
图1 眼底彩照 双眼视盘正常,右、左眼黄斑及其颞侧各可见约3×4视盘直径大小的边界清楚的椭圆形和圆形视网膜脉络膜凹陷病灶,其边缘及底部见较多色素沉着,底部见裸露之脉络膜大血管,视网膜血管在其边缘走形正常。A.右眼;B.左眼。

图2 IR眼底 双眼黄斑区清晰病灶轮廓。A.右眼;B.左眼。
频域光学相干层析成像(spectral-domain optical coherence tomography, SD-OCT)检查示双眼黄斑区巨大凹陷病灶,边缘视网膜神经感觉层与其下组织粘连紧密(图3A、B),视网膜明显萎缩,视网膜色素上皮层和脉络膜组织缺如(图3C、D)。眼科B超示双眼黄斑区挖掘样局限性凹陷,左眼病灶底部组织粘连(图4)。多焦视网膜电图(multifocal electroretinogram, mf-ERG)示双眼黄斑中心凹、旁中心凹及周边区域三维图形和轨迹阵列振幅广泛性重度降低,峰值延迟,1-5环振幅密度明显下降(图5)。双眼24-2全阈值视野检查示大范围的旁中心暗点(图6)。双眼球大小正常,各方位运动无明显受限,无眼球震颤。明确诊断:双眼先天性黄斑缺损(macular coloboma, MC)。该患者密切随访2年,未见明显眼底改变。

图3 SD-OCT检查 双眼黄斑区巨大凹陷状病灶,边缘视网膜神经感觉层与其下组织粘连紧密(A.右眼,B.左眼),视网膜明显萎缩,视网膜色素上皮层和脉络膜组织缺如(C.右眼,D.左眼)。

图4 眼科B超 双眼黄斑区挖掘样局限性凹陷,左眼病灶底部组织粘连。A.右眼;B.左眼。
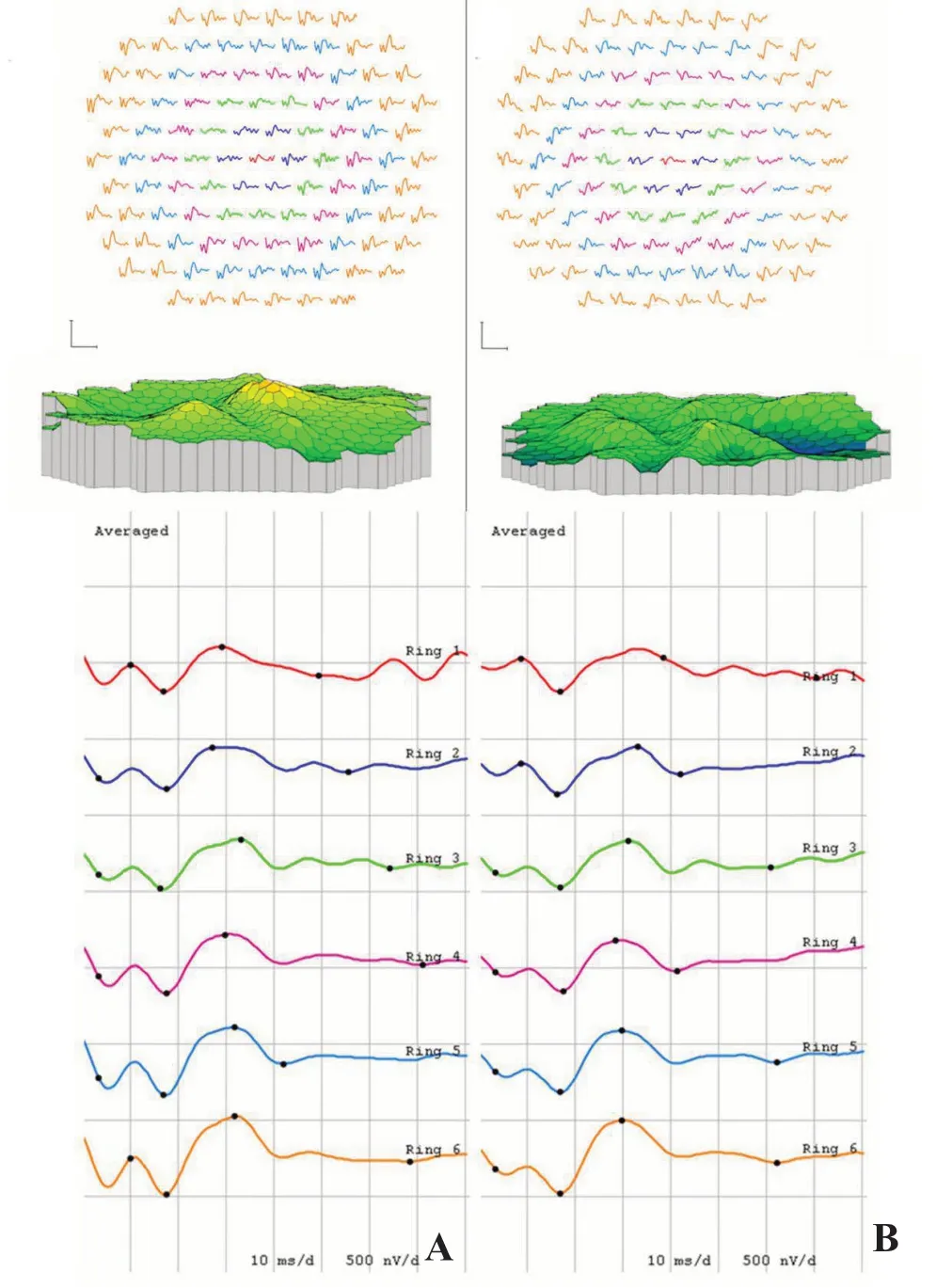
图5 mf-ERG 双眼黄斑中心凹、旁中心凹及周边区域三维图形和轨迹阵列显示振幅均广泛性重度降低,峰值延迟,1-5环振幅密度明显下降。A.右眼;B.左眼。
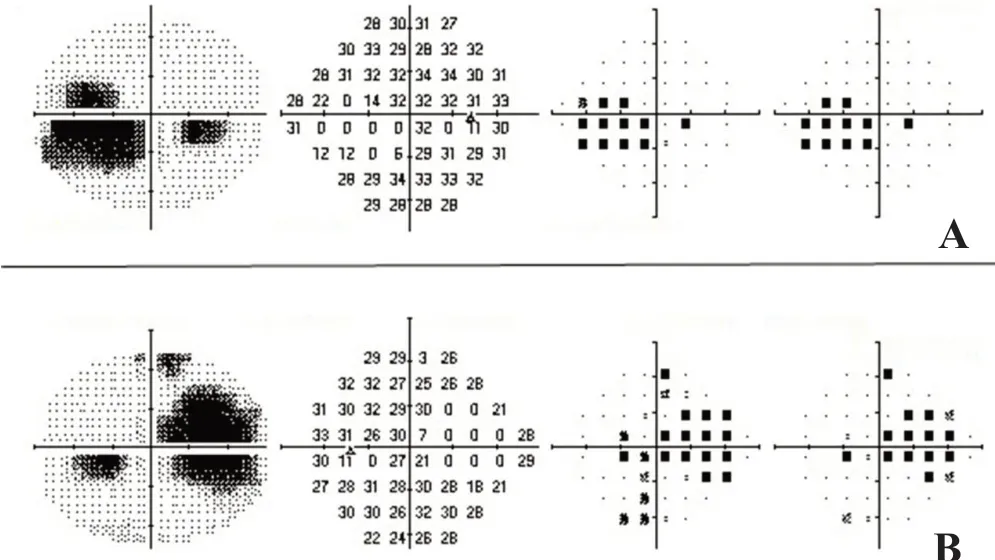
图6 双眼24-2全阈值视野 大范围的旁中心暗点。A.右眼;B.左眼。
讨论先天性MC是一种罕见的先天性黄斑区视网膜、脉络膜的发育缺陷,常严重影响患者中心视力,可单眼或双眼发病[1]。部分患者合并晶状体后囊下混浊、眼球震颤及外斜视等眼部表现,也可合并侏儒症、唐氏综合征及软骨发育不良等全身疾病[2]。其病因目前尚不明确,有学者认为与遗传有关[3-4],也有学者推测与其母亲孕期宫内感染有关[3]。
先天性MC的典型眼底表现为黄斑区边界清楚的萎缩性凹陷病灶[2]。根据Mann[5]1927年对于MC的分类,此病例应归为色素型,主要表现为黄斑区圆形或椭圆形的瘢痕样凹陷,其上覆盖密集或分布不均的色素,底部裸露脉络膜大血管。MC典型的SD-OCT表现为视网膜神经感觉层的萎缩,视网膜色素上皮和脉络膜的缺如[2,6]。MC患者的mfERG和全视野视网膜电图(full-field electroretinogram, ffERG)表现不同。MC患者ffERG中a波和b波可表现无显著异常或仅有轻度降低[7],而mfERG多表现为振幅的严重降低[3-4]。在此病例中,mfERG提示双眼黄斑中心凹及黄斑周边区域振幅均重度降低,峰值延迟。ffERG 是检测全视网膜功能,如视网膜色素变性病变可累及全视网膜,而mfERG是检测视网膜黄斑区局部病变,先天性MC属于黄斑局部病变,故其ffERG改变不大而mfERG则改变明显,说明mfERG在评估MC病情时较ffERG更具临床意义。B超检查表现为典型的黄斑区挖掘样局限性凹陷,与Lezrek等[8]的研究结果一致。双眼视野检查显示为大范围的旁中心暗点。
大多数MC患者自幼视力损害严重,多在指数~0.2之间[1]。Hou 等[2]研究发现27%的MC患者视力为0.2~0.8,其BCVA与MC类型及眼球运动异常呈正相关,而与年龄、性别、病程、MC家族史、视盘异常及存在系统性疾病无关。色素型MC视力预后最差。该患者为色素型MC,多模式影像检查均表明其黄斑功能已受到严重损害,BCVA仅为0.15。但该患者仍有中心视野,可能与其视乳头黄斑束结构尚完整,仍保留一定数量的视细胞有关[9]。
基因检测可用于研究MC患者合并眼部病变或全身疾病的发病机制,有利于该病的进一步治疗[3,10-11]。眼底荧光血管造影非临床上诊断MC的首选方法,但可辅助明确MC的分型[2]。
此外,单凭眼底彩照诊断先天性MC是稍有欠缺的。后葡萄膜炎、Stargardt病、脉络膜局限性缺损、弥漫性脉络膜营养障碍、中心性晕轮状视网膜脉络膜萎缩等疾病均可在眼底产生形态类似的病灶[8,12]。组织学上证实黄斑缺损区脉络膜毛细血管和视网膜色素上皮的缺失对于诊断MC是极为重要的,在无组织学检查的情况下,SD-OCT可详细记录和评估MC患者视网膜的超微结构来明确诊断[1]。该患者否认家族史及眼外伤史,无牛眼样改变、小眼球、视神经、虹膜及其他全身先天异常,且通过多模式影像证实黄斑区视网膜色素上皮和脉络膜缺如,可与上述疾病鉴别。综合多方因素,考虑可能为其母孕期宫内感染所致。遗憾的是,当时未做血清学检查如弓形虫、疱疹病毒及梅毒螺旋体等检查来进一步证实。
综上所述,我们应用多模式影像全面报道了1例罕见的色素型先天性MC,患者黄斑功能严重受损,视力预后较差。本研究的局限性在于病例数量和基因检测的缺乏。收集到更多的病例能够更好地阐述先天性MC的发病机制、临床表现、诊断及其治疗。
