基于高通量测序分析辣椒/玉米套作对辣椒根际土壤细菌多样性的影响
2022-05-28巩雪峰宋占锋
许 艺, 李 红, 巩雪峰, 陈 鑫, 宋占锋
(1.四川省农业科学院园艺研究所/蔬菜种质与品种创新四川省重点实验室,成都 610066;2.农业农村部西南地区园艺作物生物学与种质创制重点实验室,成都 610066)
【研究意义】辣椒/玉米套作可以提高作物水、肥、光资源利用效率,促进作物氮、磷、钾养分吸收,增加土壤微生物数量和酶活性,提高作物单位面积产量,减少作物病虫害发生,是一种很有价值的栽培模式[1-6]。目前关于此模式的研究还大量集中在栽培技术、光能利用、病虫害防控等宏观层面[4-7],相关生理生态基础理论研究还较薄弱,如土壤微生物多样性及群落组成结构研究还较少且分析方法较传统。土壤细菌是土壤微生物的重要组成成分,在土壤有机质分解、腐殖质形成、养分转化与吸收、营养元素循环等过程中起着重要作用,与土壤健康及作物生长发育和产量品质息息相关[8-9]。研究辣椒/玉米套作模式下辣椒根际土壤细菌多样性及群落组成结构,对进一步大力推广辣椒/玉米套作模式,并利用好土壤细菌具有重要指导意义。【前人研究进展】徐强等[1-3]研究了线辣椒/玉米套作对线辣椒根际和非根际土壤微生物、酶活性和土壤养分的影响及其相互关系,以及间套作玉米对线辣椒根际土壤微生物生态特征的影响,主要包括土壤微生物数量,土壤微生物量碳、氮变化,土壤微生物群落代谢剖面,土壤微生物丰富度以及土壤微生物多样性指标等,研究内容丰富全面,但研究方法主要为微生物传统分析方法——Biolog微平板法,此方法检测到的细菌群落丰度相对较低,范围较小,只能区分环境微生物总数中低于10%的微生物种类, 得到的结果不能完全反映土壤微生物多样性[10-11];康林玉等[12]利用Illumina MiSeq 2500 测序技术研究了辣椒种植前后土壤细菌和真菌的丰富度、多样性指数以及微生物群落结构变化,研究方法新颖高效,但研究对象仅为辣椒单一作物。【本研究切入点】高通量测序技术灵敏、通量高、错误率低、成本低,能简单、快速、准确地获取土壤微生物信息,真实地揭示环境中细菌群落的多样性和复杂性[8,10-12],已成为研究环境微生物群落组成和多样性的主流方法。目前,利用Illumina高通量测序技术分析辣椒/玉米套作情况下辣椒根际土壤微生物多样性的研究鲜有报道。【拟解决的关键问题】本试验拟利用Illumina高通量测序技术,分析辣椒/玉米套种模式下辣椒根际土壤细菌的多样性及其群落组成结构,从微生物角度揭示辣椒/玉米套作模式的优越性,为此模式的进一步大力推广提供基础理论依据;同时为完善此模式土壤施肥制度、改良土壤肥力、促进作物生长发育、提高栽培技术手段提供参考。
1 材料与方法
1.1 试验处理
试验地点为四川省自贡市荣县乐德镇辣椒示范园(104.40°E,29.35°N),试验共设2个处理(处理Model和CK),每个处理3次重复,共6个小区,每个小区面积100 m2。处理Model:辣椒/玉米套作—辣椒株距0.4 m,行距0.8 m;玉米定植在辣椒厢沟里面,每个厢沟定植1行,每隔2厢辣椒定植2行玉米。CK:辣椒净作—辣椒株距0.4 m,行距0.8 m。2个处理除种植方式不同外,其他农事操作均一致。
2019年11月10日,将辣椒置于穴盘育苗,2020年3月13日定植;2020年1月15日,将玉米置于穴盘育苗,2020年2月26日定植。
1.2 土壤采集及处理
在辣椒盛花盛果期(2020年6月10日)进行土壤采集处理,处理Model采集土为离套作玉米50 cm的辣椒根际土,CK采集土为辣椒根际土,土层深度20 cm。每个小区随机选择3个采样点,每个采样点采集3次土壤,然和混合均匀作为1个样品,样品编号分别为CK-1,CK-2,CK-3,Model-1,Model-2,Model-3。样品去掉土壤表层后,装入已灭菌的试管,用冰袋进行保鲜带回实验室,送罗宁生物科技有限公司进行测序。
1.3 DNA提取及测序
称取0.1 g土样,用特定DNA提取试剂盒提取DNA,再用0.8%琼脂糖凝胶电泳检测其完整性,并用紫外分光光度计检测浓度和纯度。以稀释后的基因组DNA为模板,采用16S rDNA 基因V4区域引物进行扩增,引物序列:515F(5’-GTGCCAGCMGCCGCGGTAA-3’)和806R(5’-GGACTACHVGGGTWTCTAAT-3’),扩增条件为起始94 ℃ 1 min, 然后30循环(变性94 ℃ 20 s,退火54 ℃ 30 s,延伸72 ℃ 30 s),最后72 ℃ 5 min。每个样本进行3个PCR技术重复,每个PCR反应终止于线性扩增期,PCR结束后将同一样本的PCR产物与1/6体积的6X loading buffer混合,使用2%琼脂糖凝胶电泳检测。取目的条带用来回收,回收使用QIAquick Gel Extraction Kit(QIAGEN),再用Qubit@2.0 Fluorometer(Thermo Scientific)定量,最后等摩尔量混合。使用Illumina公司TruSeq DNA PCR-Free Sample Prep Kit(FC-121-3001/3003)进行文库构建,构建好的文库经过定量和文库检测合格后,采用Hiseq 2500平台PE250模式上机测序。
1.4 数据分析
测序得到的原始数据经过FLASH8拼接双端序列,基于Barcode从reads中拆分出各样品序列,截去Barcode序列得到原始数据,然后使用Trimmomatic[13]进行质控。参考Gold数据库,使用Uchime算法[14]去除嵌合体,得到有效数据Clean Reads。基于Usearch软件,使用UPARSE算法[15]在97%的一致性水平上进行OTU聚类,挑选每个OTU中出现频数最高的序列作为OTU的代表序列。使用UCLUST分类法[16]与SILVA数据库(Rlease_123 http://www.arb-silva.de/)[17]进行注释分析。基于OTU丰度表和注释后的分类信息表,在各个分类水平对数据进行转换,分析各个样本在各个分类水平上的相对丰度;从样本中随机抽取一定量的序列,并统计它们所代表的物种数目,以抽取的一系列序列数和相应的物种数构建稀释曲线;使用R语言分析样本的Alpha-多样性指数;使用Python LEfSe包进行LEfSe分析。
2 结果与分析
2.1 稀释曲线
稀释曲线(Rarefaction Curve)可间接反映样本中物种的丰富度,由图1可知,处理Model与CK的稀释曲线均随着序列数的增加而趋于平缓,说明测序数据合理,能够比较真实地反应土壤样品的细菌群落,但尚有少量细菌种类未被发现。

图1 稀释曲线
2.2 Alpha多样性
Alpha多样性指数用Chao1、Ace、Shannon和Simpson 4个指数表示,其中Chao1和Ace指数越大,说明物种丰富度越高,即群落中物种数量越多;Simpson和Shannon指数越大,说明群落多样性越高,即个体分配越均匀。由表1可知,辣椒/玉米套作(处理Model)后,土壤细菌群落的Chao1、Ace、Shannon指数均较净作辣椒(CK)有所增加,增幅为3.07%~7.53%,说明辣椒/玉米套作后,土壤细菌群落的丰富度和多样性增加。

表1 不同处理土壤细菌丰富度和多样性指数
2.3 辣椒/玉米套作对土壤细菌群落丰度的影响
2.3.1 门水平土壤细菌优势种群 由图2可知,不同处理土壤细菌群落在门水平上排名前10的细菌种类相同,均为变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)、酸杆菌门(Acidobacteria)、厚壁菌门(Firmicutes)、芽单胞菌门(Gemmatimonadetes)、绿菌门(Chlorobi)、绿弯菌门(Chloroflexi)、浮霉菌门(Planctomycetes)、硝化螺旋菌门(Nitrospirae)和疣微菌门(Verrucomicrobia),不同处理土壤样品前10种细菌门相对丰度之和均在细菌总量的98%以上,其中变形菌门(Proteobacteria)是最大优势菌门,分别占处理Model和对照CK土壤细菌总量的88.75%和89.79%。

图2 不同处理土壤细菌在门水平上的相对丰度统计分析
对不同处理土壤样品的主要优势菌门进行分析发现,辣椒/玉米套作后土壤细菌群落组成结构发生明显变化。辣椒/玉米套作后(处理Model),土壤拟杆菌门相对丰度明显降低,降低了30.68%;酸杆菌门、芽单胞菌门、绿菌门、绿弯菌门、浮霉菌门、硝化螺旋菌门、疣微菌门相对丰度均有所增加,增幅为17.45%~67.82%,其中硝化螺旋菌门增幅为67.82%,芽单胞菌门增幅为51%。
2.3.2 纲水平土壤细菌优势种群 由图3可知,不同处理土壤细菌群落在纲水平上排名前10的细菌种类相同,均为α-变形杆菌纲(Alphaproteobacteria)、丙型变形菌纲(Gammaproteobacteria)、β-变形菌纲(Betaproteobacteria)、拟杆菌纲(Bacteroidia)、梭状芽胞杆菌纲(Clostridia)、酸杆菌纲(Acidobacteria)、芽单胞菌纲(Gemmatimonadetes)、δ-变形菌纲(Deltaproteobacteria)、ε-变形菌纲(Epsilonproteobacteria)和Ignavibacteria。处理Model和对照CK土壤样品前10种细菌纲相对丰度之和分别占细菌总量的94.97%和96.19%,其中α-变形杆菌纲是2个处理的最大优势菌纲,分别占处理Model和对照CK土壤细菌总量的75.49%和66.25%;但2个处理的次优势菌纲发生变化,对照CK的次优势菌纲为丙型变形菌纲,占细菌总量的16.44%;而辣椒/玉米套作后,处理Model的次优势菌纲变为β-变形菌纲,占细菌总量的8.77%,丙型变形菌纲变为第三优势菌纲,占细菌总量的4.40%。

图3 不同处理土壤细菌在纲水平上的相对丰度统计分析
对不同处理土壤样品的主要优势菌纲进行分析发现,辣椒/玉米套作后土壤细菌群落组成结构发生明显变化。辣椒/玉米套作后,土壤丙型变形菌纲和拟杆菌纲相对丰度明显降低,分别降低了77.47%和47.67%;β-变形菌纲、酸杆菌纲、芽单胞菌纲相对丰度明显增加,增幅分别为38.64%、40.76%、51.00%。
2.3.3 目水平土壤细菌优势种群 由图4可知,不同处理土壤细菌群落在目水平上排名前10的细菌种类相同,均为鞘脂单胞菌目(Sphingomonadales)、肠杆菌目(Enterobacteriales)、伯克氏菌目(Burkholderiales)、拟杆菌目(Bacteroidales)、亚硝化单胞菌目(Nitrosomonadales)、梭菌目(Clostridiales)、黄单胞菌目(Xanthomonadales)、芽单胞菌目(Gemmatimonadales)、根瘤菌目(Rhizobiales)和Subgroup 6。处理Model和对照CK土壤样品前10种细菌目相对丰度之和分别占细菌总量的90.57%和92.59%,其中鞘脂单胞菌目是2个处理的最大优势菌目,分别占处理Model和对照CK土壤细菌总量的74.06%和64.93%;但2个处理的次优势菌目发生变化,对照CK的次优势菌目为肠杆菌目,占细菌总量的14.85%;而辣椒/玉米套作后,处理Model的次优势菌目变为伯克氏菌目,占细菌总量的6.02%,肠杆菌目变为第三优势菌目,占细菌总量的2.05%。

图4 不同处理土壤细菌在目水平上的相对丰度统计分析
对不同处理土壤样品的主要优势菌目进行分析发现,辣椒/玉米套作后土壤细菌群落组成结构发生明显变化。辣椒/玉米套作后,肠杆菌目和拟杆菌目相对丰度明显降低,分别降低了86.19%和47.67%;伯克氏菌目、芽单胞菌目和亚硝化单胞菌目相对丰度明显增加,增幅分别为40.03%、45.30%和31.38%;其次是黄单胞菌目、鞘脂单胞菌目和Subgroup 6,增幅分别为26.98%、14.07%和13.61%。
2.3.4 科水平土壤细菌优势种群 由图5可知,不同处理土壤细菌群落在科水平上排名前10的细菌种类相同,均为鞘脂单胞菌科(Sphingomonadaceae)、肠杆菌科(Enterobacteriaceae)、丛毛单胞菌科(Comamonadaceae)、亚硝化单胞菌科(Nitrosomonadaceae)、芽单胞菌科(Gemmatimonadaceae)、毛螺菌科(Lachnospiraceae)、拟杆菌S24-7 group科(Bacteroidales S24-7 group)、螺杆菌科(Helicobacteraceae)、黄单胞菌科(Xanthomonadaceae)和紫单胞菌科(Porphyromonadaceae)。处理Model和对照CK土壤样品前10种细菌科相对丰度之和分别占细菌总量的87.07%和89.19%,其中鞘脂单胞菌科是2个处理的最大优势菌科,分别占处理Model和对照CK土壤细菌总量的73.75%和64.74%;但2个处理的次优势菌科发生变化,对照CK的次优势菌科为肠杆菌科,占细菌总量的14.85%;而辣椒/玉米套作后,处理Model的次优势菌科变为丛毛单胞菌科,占细菌总量的5.70%,肠杆菌科变为第三优势菌科,占细菌总量的2.05%。
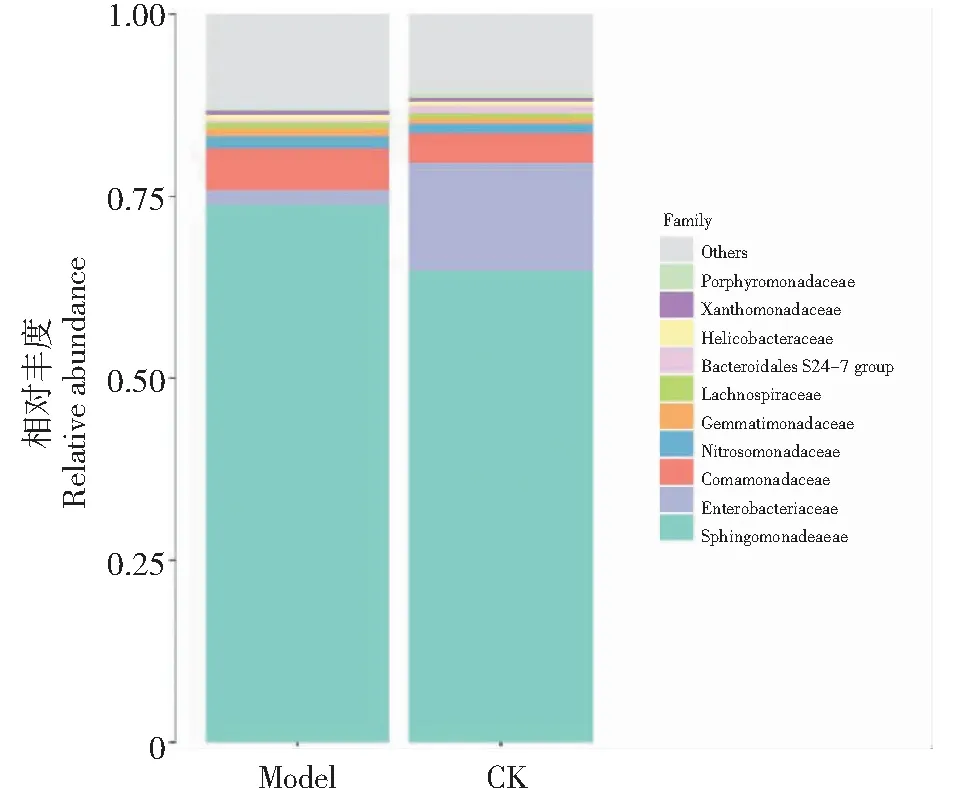
图5 不同处理土壤细菌在科水平上的相对丰度统计分析
对不同处理土壤样品的主要优势菌科进行分析发现,辣椒/玉米套作后土壤细菌群落组成结构发生明显变化。辣椒/玉米套作后,肠杆菌科和拟杆菌S24-7 group科相对丰度明显降低,分别降低了86.19%和52.64%;芽单胞菌科、丛毛单胞菌科和亚硝化单胞菌科相对丰度明显增加,增幅分别为45.30%、41.61%和31.38%;其次是黄单胞菌科和鞘脂单胞菌科,增幅分别为19.74%和13.92%。
2.3.5 属水平土壤细菌优势种群 由图6可知,不同处理土壤细菌群落在属水平上排名前10的细菌种类相同,均为鞘脂单胞菌属(Sphingomonas)、埃希氏杆菌属—志贺氏菌属(Escherichia-Shigella)、Ramlibacter、螺杆菌属(Helicobacter)、亚硝化单胞菌属(Nitrosomonas)、甲基杆菌属(Methylobacterium)、LachnospiraceaeNK4A136group、拟杆菌属(Bacteroides)、硝化螺菌属(Nitrospira)和Mucispirillum。处理Model和对照CK土壤样品前10种细菌属相对丰度之和分别占细菌总量的83.83%和86.35%,其中鞘脂单胞菌属是2个处理的最大优势菌属,分别占处理Model和对照CK土壤细菌总量的73.08%和64.69%;但2个处理的次优势菌属发生变化,对照CK的次优势菌属为埃希氏杆菌属—志贺氏菌属,占细菌总量的14.85%;而辣椒/玉米套作后,处理Model的次优势菌属变为Ramlibacter属,占细菌总量的5.26%,埃希氏杆菌属—志贺氏菌属变为第三优势菌属,占细菌总量的2.05%。

图6 不同处理土壤细菌在属水平上的相对丰度统计分析
对不同处理土壤样品的主要优势菌属进行分析发现,辣椒/玉米套作后土壤细菌群落组成结构发生明显变化。辣椒/玉米套作后,埃希氏杆菌属—志贺氏菌属和拟杆菌属相对丰度明显降低,分别降低了86.19%和53.18%;Ramlibacter属和硝化螺旋菌属相对丰度明显增加,增幅分别为34.85%和105.82%;其次是鞘脂单胞菌属,增幅为13.89%。
2.4 差异物种LEfSe分析
LEfSe(LDA Effect Size)分析可以找到重要的、在组间有显著差异的物种,其统计结果包括3部分,其中LDA值分布柱状图展示每个组内显著富集的物种及其重要性程度,物种分支进化图展示差异物种及其进化关系。由图7~8可知,处理Model中具有显著性差异的细菌种群共56种,显著差异物种影响大小(重要性)排名前5的依次是亚硝化单胞菌科、酸杆菌纲Subgroup_4目、亚硝化单胞菌目、硝化螺旋菌门、RB41科;CK中具有显著性差异的种群共10种,显著差异物种影响大小(重要性)排名前5的依次是假单胞菌目、不动杆菌属、脱铁杆菌目、莫拉氏菌科和脱铁杆菌纲。处理Model中显著富集并达到组间显著性差异的微生物物种明显多于CK。
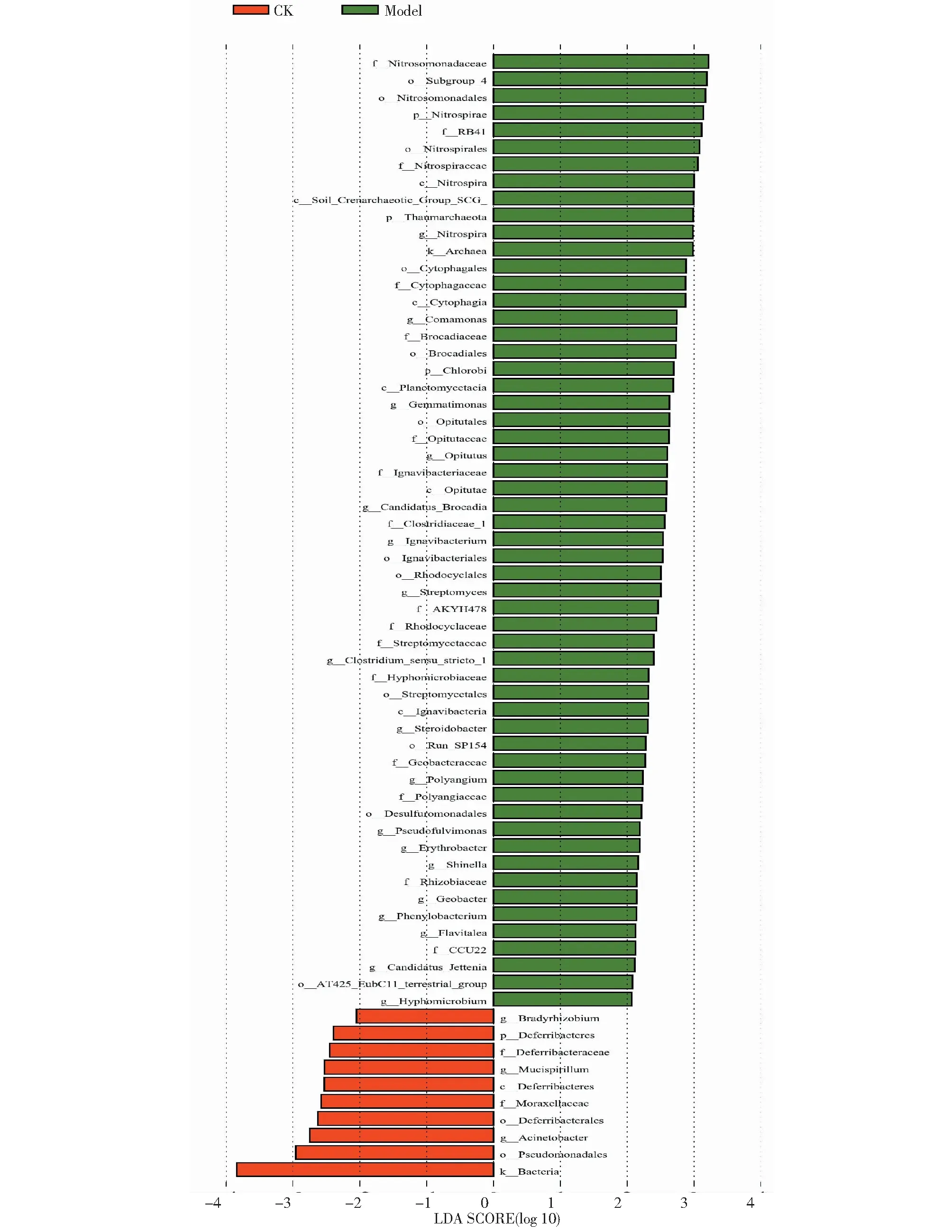
图7 LDA值分布
在处理Model中,富集微生物达到显著差异水平且有进化关系的细菌物种主要有亚硝化单胞菌目、亚硝化单胞菌科;硝化螺旋菌门、硝化螺旋菌纲、硝化螺旋菌目、硝化螺旋菌科、硝化螺旋菌属;绿菌门、Ignavibacteria纲、Ignavibacteriales目、Ignavibacteriaceae科;丰佑菌纲、丰佑菌目、丰佑菌科、丰佑菌属。在对照CK中,微生物达到显著差异水平且有进化关系的细菌物种主要有脱铁杆菌门、脱铁杆菌纲、脱铁杆菌目、脱铁杆菌科。

图8 物种分支进化图
3 讨 论
土壤细菌是土壤微生物的重要组成成分,对土壤营养元素循环、有机质形成与分解、肥力保持,以及植物养分吸收与生长发育等均有重要促进作用,其多样性及群落组成结构受环境因子、土壤特性、植物种类与不同种植方式等多种因素影响[1,8,18]。套作是植物生产中常见的种植方式,其直接增加了植物群落多样性,Cartwright和Fierer等[19-20]认为土壤微生物多样性与植物群落多样性呈正相关。本试验中,辣椒/玉米套作后土壤细菌群落的Chao1、Ace、Shannon指数增加,土壤细菌群落的丰富度和多样性增加,与前人研究结果[1]一致。套作后作物根际土壤细菌多样性增加可能是因为套作影响了土壤温度、湿度等环境因子,同时改变了作物生长的土壤微生态环境,如根系分泌物、作物残体、根系残留物等的积累促进了土壤微生物活动,从而提高了土壤微生物的群落结构多样性[1-2,12]。徐强等[1]研究表明,线辣椒根际土壤微生物的不同多样性指数分别与其生物学产量之间存在显著或极显著正相关,说明土壤微生物多样性增加是辣椒/玉米套作后产量增加的重要因素之一。
本试验中,辣椒/玉米套作与辣椒净作的辣椒根际土壤细菌群落在门、纲、目、科、属水平上排名前10的细菌种类均相同,说明相同作物的土壤细菌微生物群落具有一定的稳定性。其中变形菌门是细菌域中最大的一门,多为革兰氏阴性菌,其种类繁多且生态功能多样,对环境有极强的适应性和变异性,其中许多细菌有固氮作用,可增加土壤中的氮素营养,促进植株生长,是多种生态系统的最大优势菌种[8-9,21-22]。本试验中辣椒/玉米套作与辣椒净作的最大优势菌门也是变形菌门,均占土壤细菌总量的88%以上。但Heiko等[23]发现欧洲山毛榉和挪威云杉根际土壤细菌类群的最大优势菌种为酸杆菌;Fierer等[24]也发现在温带森林中酸杆菌是土壤中含量最丰富的细菌类群;而魏鹏等[25]发现放线菌门在南缘伊犁绢蒿荒漠、腹地白梭梭荒漠和北缘盐生假木贼荒漠3种荒漠类型中相对丰度最高,说明不同生态系统存在不同的优势菌种。
硝化作用是硝化细菌利用二氧化碳为碳源,将铵态氮氧化为亚硝酸盐继而氧化为硝酸盐,并从中获取能量的微生物过程。硝化作用的发生消耗了土壤和外源肥料中的铵态氮,减少了氨挥发损失,是土壤氮素循环的核心和提供植物有效氮的主要过程[26-27]。亚硝化单胞菌和硝化螺旋菌是参与土壤硝化作用的2个重要菌群[28]。研究[29]表明,在硝化菌中,硝化螺旋菌有较强的代谢活性,对底物亲和力较强,在低浓度氨氮环境中竞争优势明显。本试验通过差异物种LEfSe分析发现,辣椒/玉米套作后,辣椒根际土壤亚硝化单胞菌和硝化螺旋菌显著提高,说明辣椒/玉米套种后辣椒根际土壤硝化作用更加活跃,氮素循环增强,进而可能有利于辣椒根际土壤氮养分的释放和有效化,提高作物氮元素的吸收量和利用率。徐强等[30]研究结果也表明,线辣椒/玉米套作后其氮素吸收量超过了相应作物单作时的吸收量,也高于单作吸收量按套作比例加权的平均吸收量,表现出氮素吸收套作优势。但本研究仅分析了作物根际微生物情况、微生物群落结构变化与土壤养分的相关性,、以及植物营养元素吸收利用机理等还需进一步试验研究。
4 结 论
辣椒/玉米套作后,辣椒根际土壤细菌群落的Chao1、Ace、Shannon指数增加,丰富度和多样性增加;通过土壤细菌群落丰度分析发现,2个处理在门、纲、目、科、属水平上排名前10的优势细菌种类相同,最大优势菌种分别为变形菌门、α-变形杆菌纲、鞘脂单胞菌目、鞘脂单胞菌科和鞘脂单胞菌属,但各细菌数量发生变化,导致土壤细菌群落组成结构发生变化;通过差异物种LEfSe分析发现,辣椒/玉米套作后,辣椒根际土壤细菌富集并达到组间显著性差异的微生物种类有56种,较辣椒净作明显增加,其中亚硝化单胞菌科(从目到科)、硝化螺旋菌属(从门到属)、Ignavibacteriaceae(从门到科)、丰佑菌属(从纲到属)显著增加,脱铁杆菌科(从门到科)显著减少。
