基于计算机模拟技术从中药天然产物库中挖掘色氨酸羟化酶1抑制剂
2022-05-24史海龙冯雪松王月雯
史海龙,程 怡,黄 月,冯雪松,王月雯,黄 峰,晁 旭
基于计算机模拟技术从中药天然产物库中挖掘色氨酸羟化酶1抑制剂
史海龙,程 怡,黄 月,冯雪松,王月雯,黄 峰,晁 旭*
陕西中医药大学,陕西 西安 712046
基于计算机模拟技术从中药天然产物库中挖掘色氨酸羟化酶1(tryptophan hydroxylase-1,TPH1)抑制剂。搭建分子对接、类药性筛选、药动学预测、分子动力学模拟于一体的药物筛选平台,从中药系统药理学数据库与分析平台(traditional chinese medicine systems pharmacology database and analysis platform,TCMSP)数据库中挖掘TPH1抑制剂。川贝酮碱(TCMSP_ID:MOL009572)实验数据均表现出良好的TPH1抑制活性及类药性,预测命中分子可有效抑制胃肠道相关TPH1活性,但对中枢神经系统相关TPH2活性抑制作用较小;通过酶活抑制实验验证抑制效应,并借助分子动力学模拟动态解析结合能变化及各项能量分布。整合多种虚拟筛选技术挖掘到缓解肠易激综合征相关痛症的TPH1抑制剂-川贝酮碱MOL009572。
分子对接;分子动力学模拟;色氨酸羟化酶;抑制剂;川贝酮碱
5-羟色氨酸(5-hydroxytryptophan,5-HT)信号通路在内脏痛觉高敏感、肠动力异常等效应产生中扮演着重要角色[1],科学家们期待以此为突破口,寻求针对肠易激综合征、溃疡性结肠炎等胃肠道疾病的治疗策略。色氨酸羟化酶(tryptophan hydroxylase,TPH)可以使色氨酸的羟基化,使色氨酸转化为5-HT。这是5-HT生物合成过程中的起始步及限速步,因此TPH是治疗或缓解5-HT相关肠易激综合征(irritable bowel syndrome,IBS)痛症药物的关键靶标[2]。哺乳动物TPH普遍存在2种亚型,其中人类TPH1与TPH2基因分别位于11、12号染色体上,序列相似性高达71%[3]。TPH1在胃肠道等消化道上皮组织中表达,TPH2主要在中枢神经系统的神经细胞中表达[4],因此利用TPH1表达的组织特异性研发抑制剂,阻断消化系统内5-HT合成,可发挥IBS镇痛止痛的疗效。目前已有TPH1抑制剂类药物上市,但临床用药中仍然存在诸多问题,如长期服用可能导致毒副作用甚至耐药性,或引发脑部中枢5-HT合成阻断[5],因此需要研发新型TPH1抑制剂。
近几年研究相继解析出高分辨率TPH蛋白晶体结构,推动了基于靶标活性口袋结构的计算机辅助药物分子设计,然而国内外IBS适应症药物研发主要围绕着TPH1为靶标开发新型单一抑制剂/拮抗剂,未涉及到TPH2酶活抑制的负效应。因此本研究搭建类药性筛选、药动学参数预测、双靶标分子对接、分子动力学模拟于一体的药物筛选平台,从中药天然产物库中挖掘TPH1抑制剂。
1 材料与计算方法
分子对接均采用Schrodinger 2018的Glide模块、ADME参数计算采用Schrodinger 2018的QikProp模块,隐性溶剂环境的结合自由能计算采用分子力学/广义波恩表面积模型MM/GBSA[6],显性溶剂环境的结合自由能计算采用分子力场/泊松玻尔兹曼表面积模型MM/PBSA[7],分子动力学模拟采用Gromacs 2019与Gaussian 09程序包。
人源性的重组表达TPH1、TPH2由陕西中医药大学针药结合省级重点实验室朱先伟博士提供;LX-1031(CAS 945976-76-1,批号637958)购于abcam中国上海分公司;川贝酮碱(CAS 103530-47-8,批号20190718)购于上海同田生物;ELX808酶标仪(美国伯腾公司)。
1.1 靶蛋白TPH1/TPH2结构预处理与对接口袋分析
首先从PDB数据库下载的靶蛋白晶体结构,TPH1选择3HF8(PDB_ID),TPH2选择4V06(PDB_ID),见图1;然后采用Schrodinger的Protein Preparation Wizard模块预处理受体蛋白结构;随后采用Protein Grid Generation模块准备靶标TPH1/TPH2蛋白活性口袋并产生格点文件,原结晶配体坐标作为格点盒子中心。由于篇幅限制,仅展示靶标TPH1与原共结晶配体LP533401结合模式及关键残基,见图2。

图1 靶标受体蛋白TPH1和TPH2活性口袋
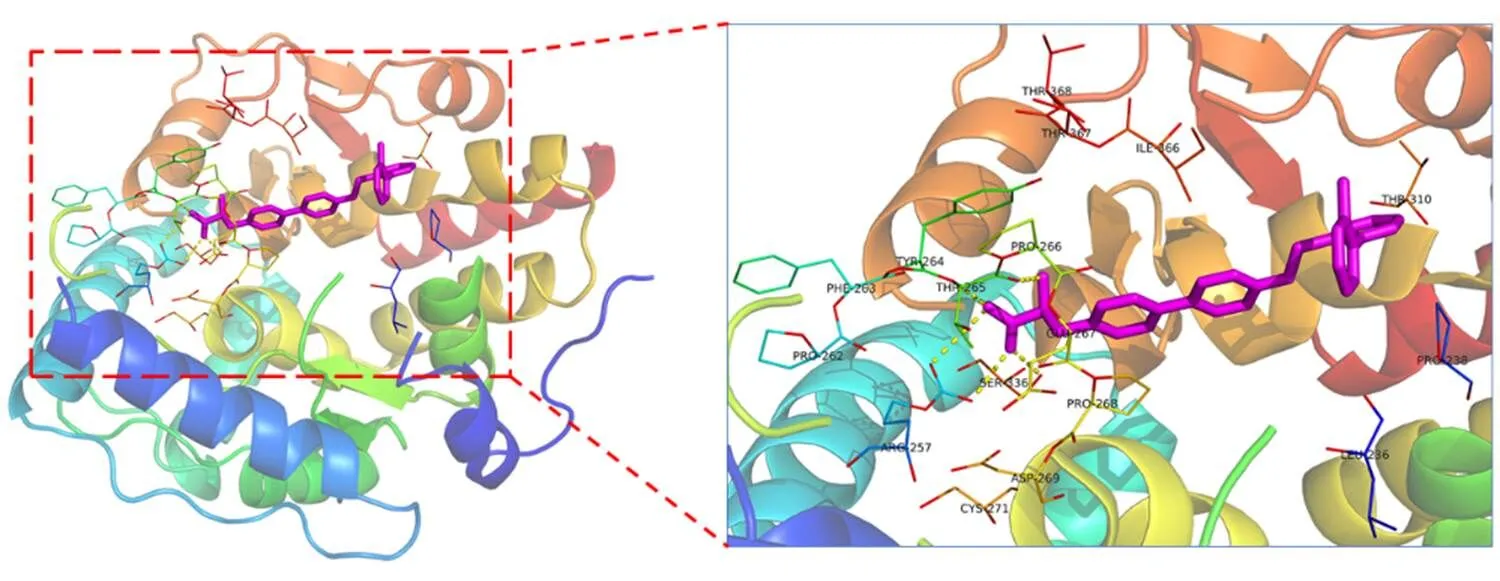
图2 晶体复合物3HF8中TPH1与原配体LP-533401结合模式
1.2 中药分子化学结构的预处理
中药天然产物数据库TCMSP[8](https:// old.tcmsp-e.com/index.php)13 144个化合物作为配体库。采用Schrodinger的LigPrep模块对各分子进行加氢、能量最小化、几何优化等处理。在力场OPLS3下,计算pH为7.0±2.0的化合物离子状态,搜索合理构象作为对接起始构象,形成可能的互变异构体。由于LX-1031有效抑制TPH1酶活性,因此可作为阳性对照,用于5-HT过表达相关疾病的药物研发,如腹泻型IBS[9]。以同样方式预处理LP533401、LX-1031,作为本实验对照组。
1.3 基于类药性评估、分子对接、ADME预测、结合自由能(隐式溶剂模型)计算的多轮筛选
基于类药性规则“Lipinski Ro5[10]”和“Verber Ro3[11]”对中药分子配体库进行首轮筛选,挑选出8987个符合类药性规则的命中分子。
基于受体结构的双靶标TPH1/TPH2分子对接进行第2轮筛选。首先针对靶标TPH1对上一步筛选的化合物库进行高通量虚拟筛选(high throughput virtual screening docking,HTVS- docking),并根据Glide Score对HTVS-docking的输出化合物进行排名。然后提取最高得分前35%化合物,进行靶标TPH1标准精度(standard precision docking,SP-docking)模式分子对接,根据Glide Score对SP-docking的输出化合物再次进行排名。随后提取最高得分前40%化合物,进行TPH1/TPH2双靶标高精度(extra precision docking,XP-docking)模式分子对接,以TPH1的Docking score≤−33.49 kJ/mol为筛选阈值,选取与TPH1有较低结合自由能的分子,再次选取与TPH2有较高Docking score的化合物,二者交集最终作为候选分子。
基于ADME药动学参数,采用Schrodinger的QikProp模块进行第3轮筛选,从相对分子质量(molecular weight,W)、氢键供体(hydrogen bond donor,HB donor)、氢键配体(hydrogen bond acceptor,HB acceptor)、溶剂可及表面积(solvent accessible surface area,SASA)、K+通道阻断相关log IC50系数(log IC50for blockage of HERG K+channels,logHERG)、水溶性系数(aqueous solubility,logS)、脂水分配系数[octanol/water partition coefficient,log(O/W)]、口服生物利用度(oral absorption,OB)、化合物的极性表面积(PSA,Van der Waals surface area of polar nitrogen and oxygen atoms and carbonyl carbon atoms)等评估,筛选具有良好类药性质的化合物。具体筛选规则为W可接受范围小于650、HB donor数值可接受范围不大于5、HB acceptor数值可接受范围不大于10、SASA数值可接受范围为300~1000、K+通道阻断相关log IC50(数值推荐区间低于-5)、logS数值可接受范围为−7.0~0.5)、log(O/W)数值可接受范围为−2~6.5、OB低于50代表较差,高于80代表较好,PSA数值可接受范围为7.0~200.0。
基于Prime-MM/GBSA结合自由能预测进行第4轮筛选。由于高精度分子对接函数计算仍然无法估算配体-受体复合物结合的溶剂化效应因素,因此使用隐性溶剂模型下MM/GBSA算法优化排序,进一步提高结合自由能的计算精度。
1.4 分子动力学模拟与MM/PBSA结合自由能(显式溶剂模型)
为了从动力学和热力学角度动态分析靶标TPH1与命中分子互作模式,使用GROMACS进行分子动力学模拟。蛋白拓扑参数取自Amber的FF03力场,由PDB2GMX工具获得;抑制剂的各原子电荷先用Gaussian 09在B3LYP/6-311G**基组水平计算静电势,再用Amber中的RESP电荷拟合程序算出。将复合物放入1 nm×1 nm×1 nm的立方体TIP3P水盒中,并加入Na+和Cl−,中和该系统到0.15 mol/L NaCl溶液浓度,随后进行能量最小化和预平衡,最后在26.85 ℃(300 K)进行50 ns的分子动力学模拟,时间间隔为2 fs,每10 ps保存一次能量和坐标文件。从25~50 ns稳定模拟轨迹中提取250帧轨迹文件,计算MM/PBSA结合自由能,并且提取TPH1- MOL009572、TPH1-LX-1031两个体系分子动力学模拟50 ns最后一帧构象,解析受体-配体结合模式。
1.5 TPH酶活抑制测定
命中分子MOL009572进行TPH酶活抑制的验证性实验。酶活测定缓冲液包含:60 μmol/L色氨酸溶液、300 mol/L 6-甲基-四氢蝶呤、200 mmol/L硫酸铵溶液、7 mmol/L DTT、25 μg/mL过氧化氢酶、25 μmol/L硫酸亚铁铵、50 mmol/L MES缓冲液(pH 7.0)。25 nmol/L人源性TPH1/TPH2被添加到不同浓度梯度的MOL009572溶液中开始反应。使用4 mm的石英比色皿荧光分光光度法定量测定酶活性,激发光波长为300 nm,发射光波长为330 nm,分别于狭缝宽度在3 nm处激发和5 nm处发射。
2 结果与分析
2.1 虚拟筛选结果分析
TCMSP数据库中13 144个中药单体化合物进行多轮虚拟筛选,筛选流程见图3,挑选出排名前10的命中分子,它们均具备有较强的TPH1结合自由能、较弱的TPH2结合自由能、良好的药动学参数、较高的OB等,如表1、2所示。进一步参照LX-1031的Prime-MM/GBSA结合自由能,以−57.73 kJ/mol作为阈值,筛选得到2个抑制剂分子、MOL009572(川贝酮碱,chuanbeinone)、MOL009586(异浙贝母碱,isoverticine),二者均是来源于川贝母的等甾体生物碱,结构式见图4。
2.2 分子动力学模拟轨迹的均方根偏差(root-mean- square deviation,RMSD)及结合自由能计算
计算分子动力学模拟50 ns时长轨迹的RMSD值,以此检验体系稳定性,结果见图5。在模拟运行15 ns之后,所有体系的RMSD值均无大幅波动,均达到了平衡状态。从25~50 ns分子动力学轨迹中提取250 帧轨迹文件,计算MM/PBSA结合自由能及各种能量贡献情况,结果见表3。结合自由能强弱关系为MOL009572>LX-1031>MOL009586>LP533401,其中范德华力最有利于命中分子与靶标结合,并起主导作用;静电势能、表面溶剂化作用也有利于结合,但作用力较弱,而溶剂中极性溶剂化作用不利于体系结合。在LP-533401与靶标结合中,静电势能、范德华力均有利于配体和受体的结合,静电势能处于主导地位;但是极性溶剂化作用力较强,不利于体系结合;在候选分子MOL009586中,与靶标的范德华力较弱,而溶剂中的极性相互作用较高,2个因素均不利于体系结合,造成与TPH1结合自由能数值偏高。基于以上分析,选择MOL009572作为TPH1的抑制分子。
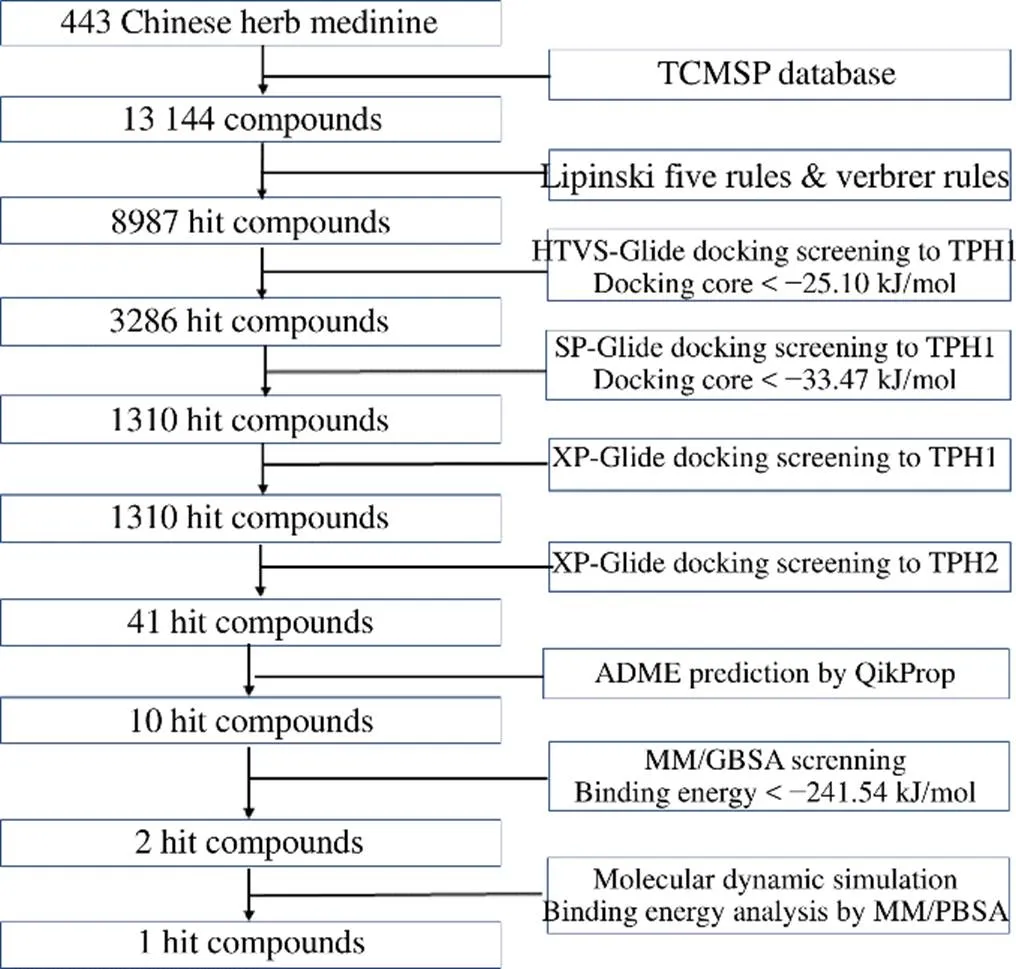
图3 虚拟筛选流程及结果

表1 命中分子(排名前10) 双靶标TPH1/TPH2对接结合能及MM/GBSA结合自由能
2.3 分子动力学模拟轨迹RMSD值解析
计算15~50 ns TPH1蛋白骨架原子的均方根波动值(root mean square fluctuation,RMSF),结果见图6。体系TPH1-MOL009572、TPH1-LX-1031中蛋白骨架原子波动变化趋势相似,氨基酸残基222~225区段和361~371区段出现较大波动,无法形成稳定的成键相互作用,不利于受体-配体复合物的结合,但是2个体系的整体性波动幅度均小于空载体蛋白靶标,说明MOL009572、LX-1031与靶标的结合,有利于靶标蛋白空间结构稳定。除此之外,还观察到2个体系在氨基酸残基270~300区段、310~320区段出现了较小波动(RMSF<0.1 nm),TPH1活性口袋周围氨基酸残基(Leu236、Pro238、Arg257、Pro262、Phe263、Tyr264、Thr265、Pro266,Glu267、Pro268、Asp269、Cys271、Thr310、Ile366、Thr367、Thr368,见表4)RMSD波动值均小于空受体蛋白APO、LX-1031相应的残基位点,推测MOL009572与TPH1之间形成了氢键、疏水相互作用、π-π堆积作用等,将MOL009572包裹在蛋白活性口袋内,形成更稳定的受体-配体二元复合物。
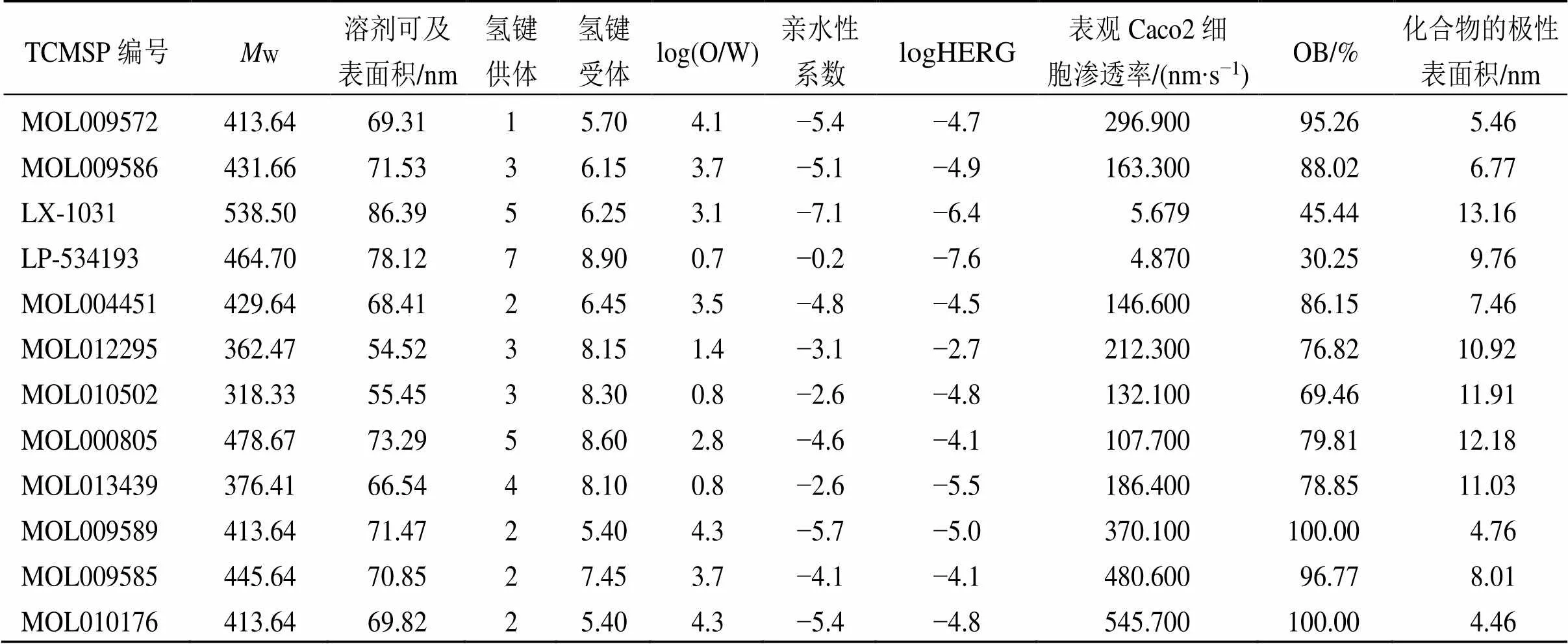
表2 命中分子(排名前10)的ADME参数预测值
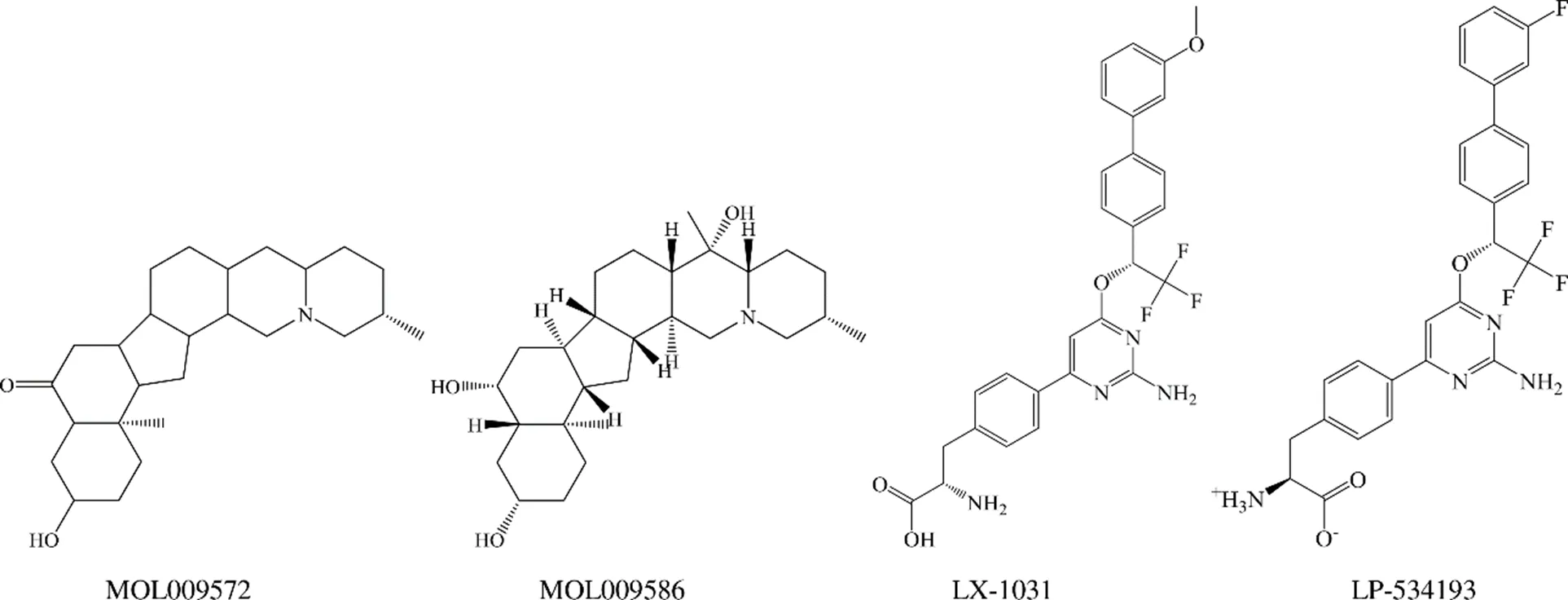
图4 命中分子和阳性对照分子的化学结构
2.4 酶活抑制实验结果
如图7所示,分别为MOL009572对酶TPH1、TPH2活性抑制实验和LX-1031对酶TPH1、TPH2活性抑制实验,这4组实验数据均由酶动力学实验方法测量得到。图7-a的实验数据计算得出,MOL009572与TPH1、LX-1031与TPH1二者的体外IC50相同,均为210 μmol/L。图7-b的实验数据计算得出,LX-1031与TPH2的体外IC50160 μmol/L,MOL009572与TPH2未观察到抑制效果。需要提出说明的是,以上每个浓度均做3次平行重复,分别取酶抑制活性平均值。此外从图7-c实验数据作Line-weaver-Burk曲线,结果表明MOL009572显示出竞争性抑制的特点。
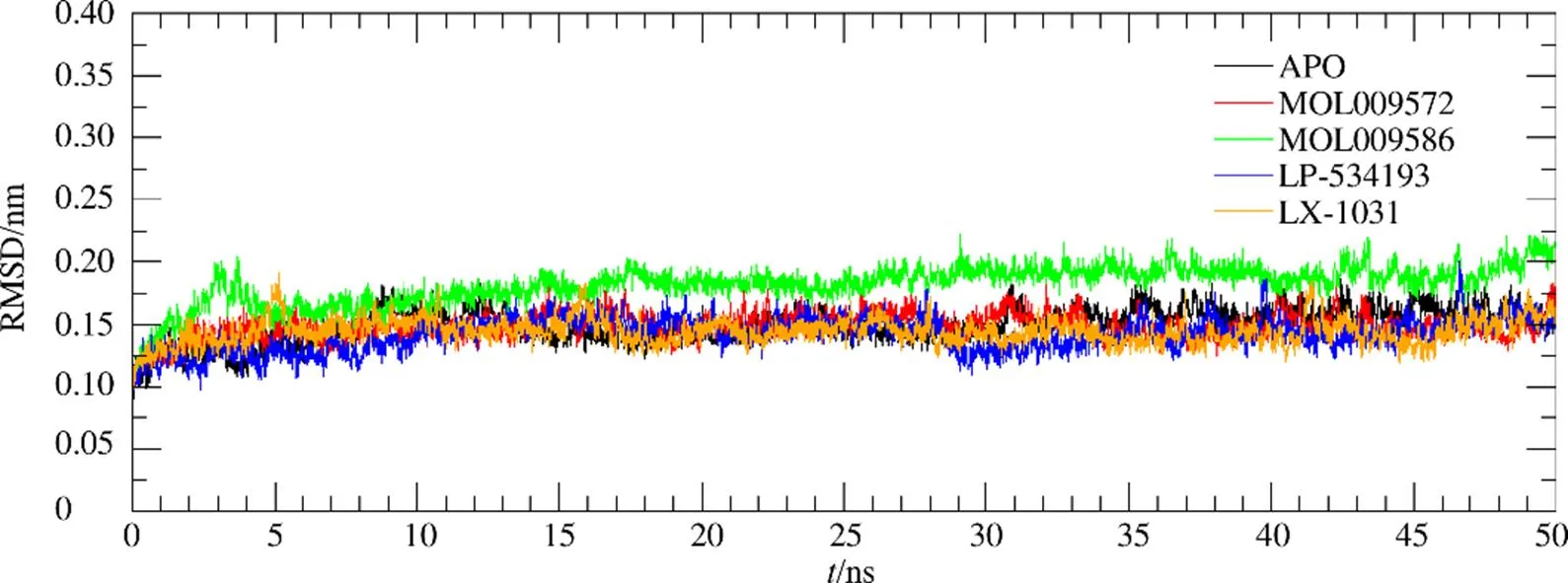
图5 分子动力学模拟过程中复合物的TPH1氨基酸骨架原子随时间变化的RMSD值

表3 MM/PBSA结合自由能
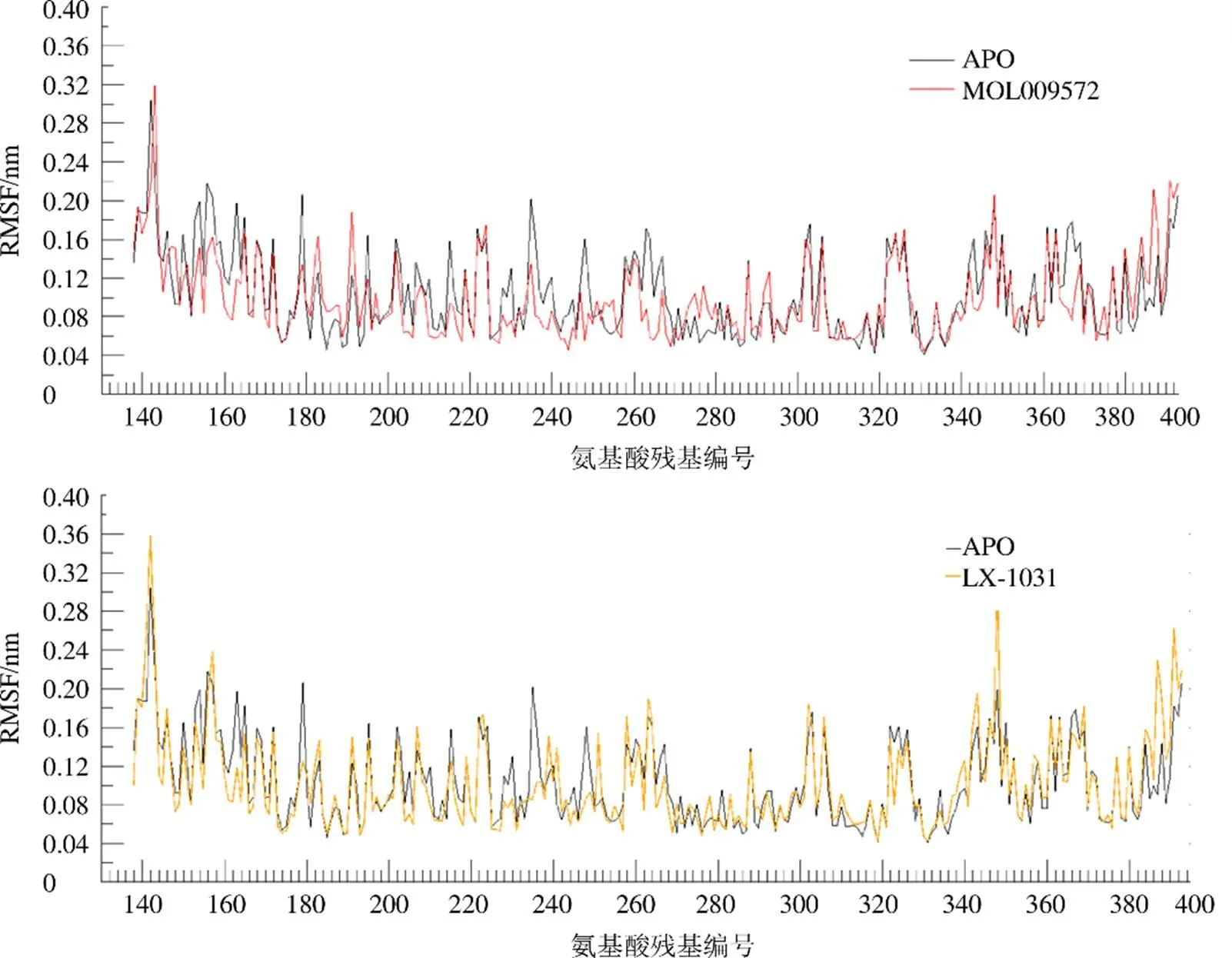
图6 分子动力学模拟过程中复合物的TPH1氨基酸骨架原子随时间变化的RMSF值
2.5 结合模式分析及分子动力学模拟氢键数目解析
Discovery Studio Visualizer分析MOL009572与TPH1互作的主要作用力是氢键、π-Alkyl堆积作用,见图8。MOL009572与氨基酸残基Tyr264形成常规氢键,与Thr265形成碳氢键,并且与在Val232、Leu236、Pro268形成Alkyl相互作用,与Phe241、Tyr235、Phe318、His272形成π-Alkyl堆积作用,这些作用力促进受体-配体相互结合。如图9所示,TPH1与LX-1031的相互作用不仅有氢键,还有π-π堆积作用。LX-1031与残基Glu317、Ser336、Arg257、Thr265形成常规氢键,与残基Tyr235、Arg121形成碳氢键,同时还与Tyr235、Phe313、His272形成π-π堆积作用,与Ile366、Pro268形成π-Alkyl或Alkyl相互作用。这些作用力使结合能降低,LX-1031亲和力增加,与TPH1活性部位紧密结合,形成稳定的受体-抑制剂复合物,降低TPH1酶的催化活性,从而有效抑制5-HT合成。
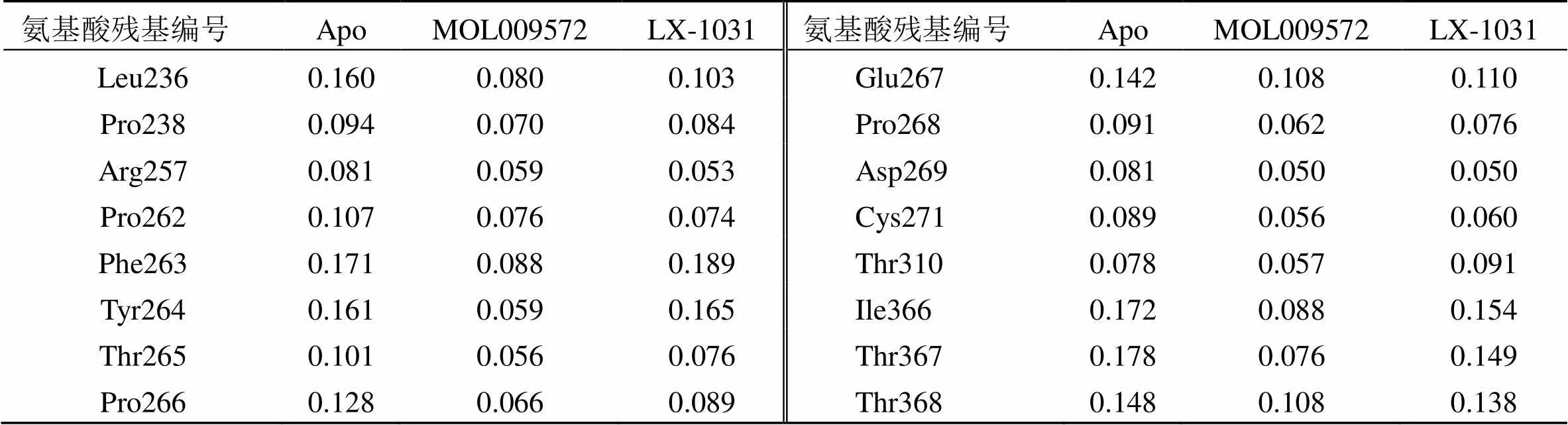
表4 分子动力学模拟过程中TPH1活性口袋部位关键残基RMSF值
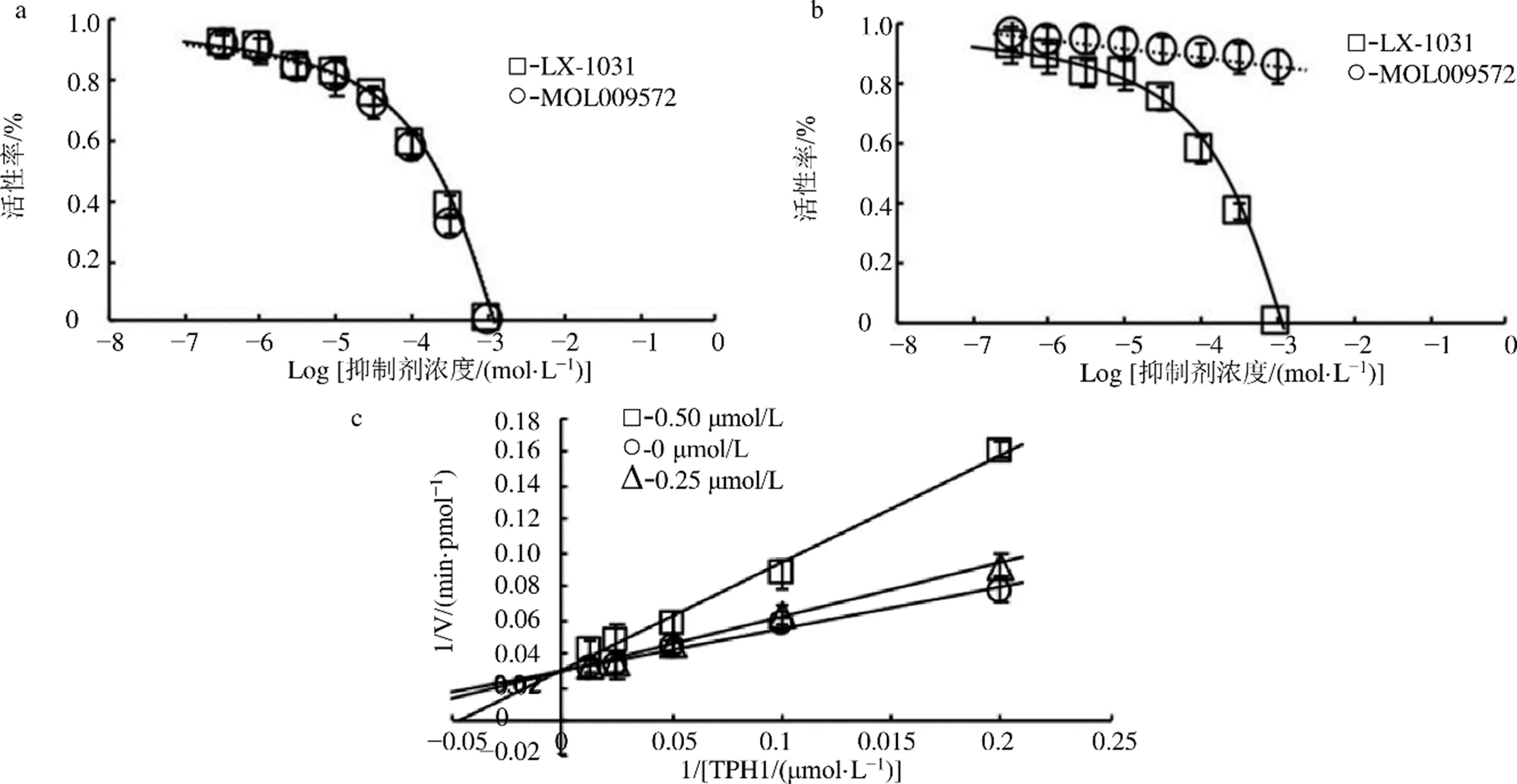
a-对人源性TPH1的抑制效果 b-对人源性TPH2的抑制效果 c-不同浓度抑制剂MOL009572的初速度的Lineweaver-Burk曲线
借助g-hbond程序计算TPH1与MOL009572、LX-1031之间的氢键数量随时间的变化,统计结果如图10所示。总体而言,与LX-1031相比,MOL009572与TPH1结合氢键数量近似;其中MOL009572氢键数量峰值为3,均值为3;LX-1031氢键数量峰值为4,均值为2,说明氢键促使复合物形成稳定构象。在分子动力学模拟45 ns之后,LX-1031与TPH1至少存在3次氢键相互作用的接触,而LX-1031与TPH1仅存在至少2次氢键相互作用的接触,提示MOL009572比LX-1031与靶标TPH1结合更具优势。
3 讨论
四川大学华西医学院何菱研究团队等使用分子动力学模拟、结合自由能预测、能量项拆分及丙氨酸扫描技术,研究TPH1抑制剂苯丙氨酸衍生物的抑制作用机制[12]。结果表明,萘取代基的同分异构体可以与靶标形成不同的结合模式,但是导致相同酶活抑制效果。随后该团队使用基于配体结构的阐明一系列已知TPH1抑制剂的定量构效关系,并提出多元药效团模型的研究策略[13],即整合多种TPH1抑制剂复合物晶体结构构建药效团模型。该模型成功预测了32种苯丙氨酸取代基衍生物的酶活抑制常数。目前国内外内脏痛觉高敏感的药物研发策略大体类似于上述案例,主要考虑TPH1单一靶标的抑制剂,未涉及到TPH2酶活抑制引起的负效应,筛选策略均较为单一,预测精度有待提升。

图8 TPH1与MOL009572相互作用
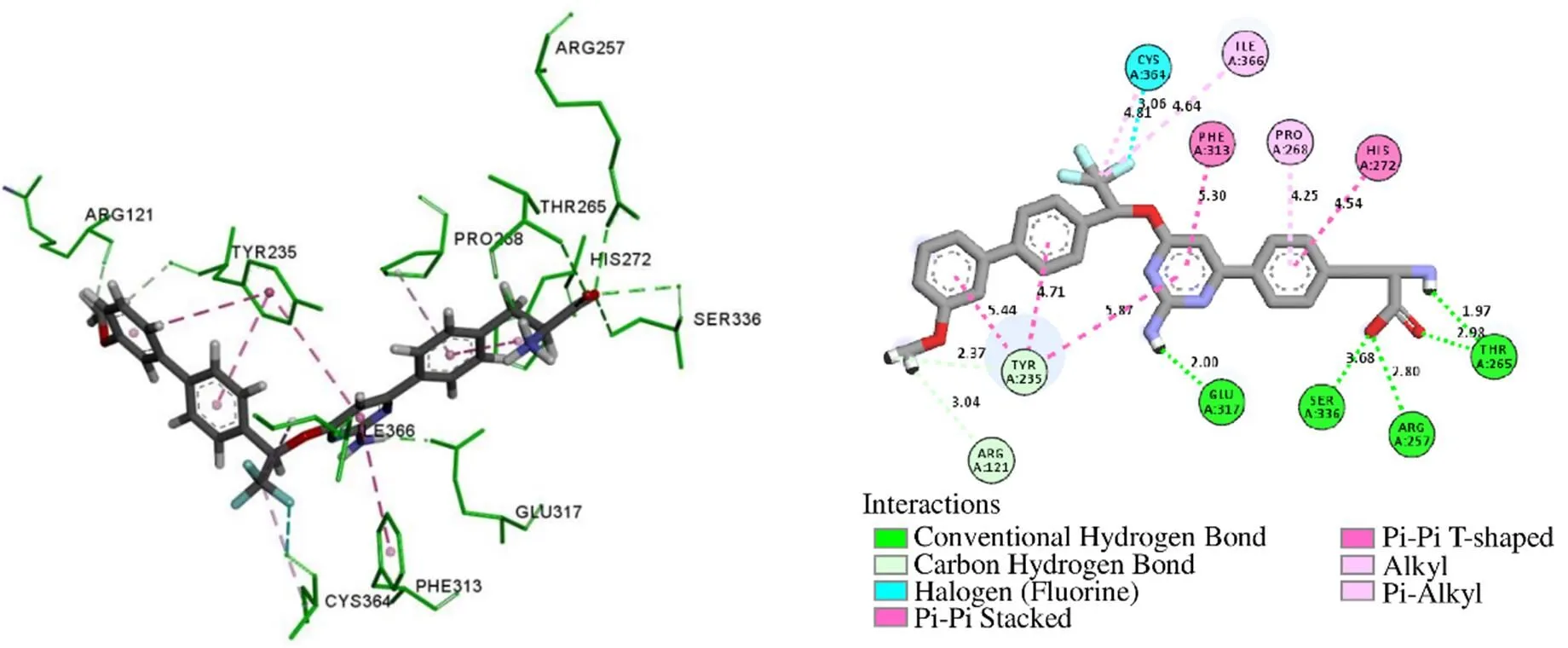
图9 TPH1与LX-1031相互作用
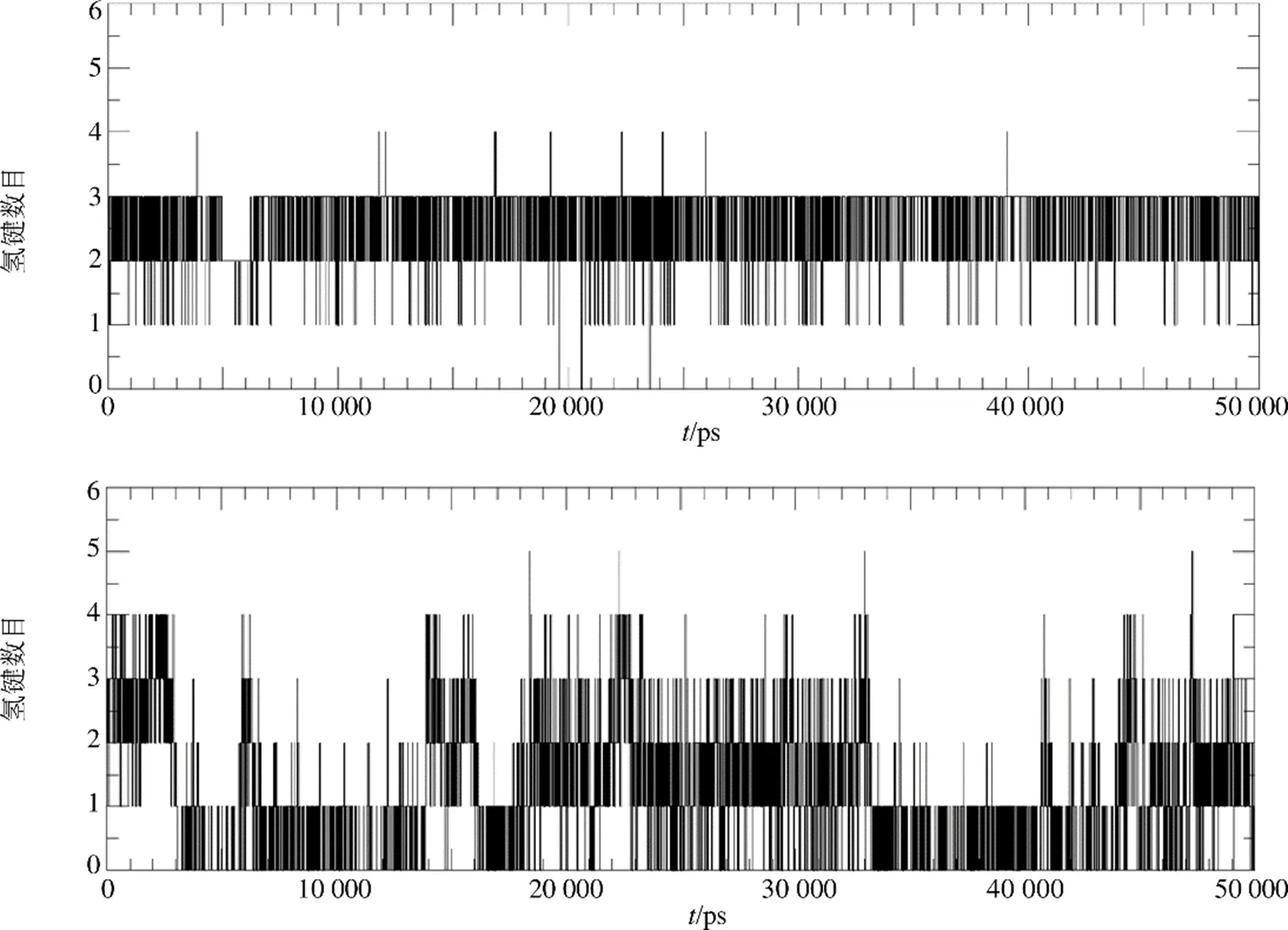
图10 分子动力学模拟过程中配体小分子与受体蛋白活性口袋形成的氢键数目
基于上述考虑,本研究提高从挖掘传统中药库药效成分为突破口,制定了一套整合类药性筛选、药动学参数预测、双靶标分子对接、结合自由计算的研究策略;对TCMSP数据库进行系统筛选,找到一种来源于中药川贝母的川贝酮碱(chuanbeinone,TCMSP-ID:MOL009586),随后进行酶活抑制实验验证,初步证明其可抑制胃肠道相关TPH1酶活性,并降低了中枢神经系统相关TPH2酶活抑制的负效应。最后采用分子动力学模拟技术从原子水平上深度剖析命中分子与靶标TPH1相互作用方式,并且分别在隐性溶剂环境下和显性溶剂环境下计算结合自由能。本研究挖掘IBS相关痛症的中药药效分子,可为研发新型TPH1抑制剂提供参考。然而,本研究还处于药物开发的前期阶段,尚缺乏药动学评估等实验数据支撑,后期将联合实验药理学团队,开展相关生物学活性验证及安全性评价。
利益冲突 所有作者均声明不存在利益冲突
[1] Okaty B W, Commons K G, Dymecki S M.Embracing diversity in the 5-HT neuronal system [J]., 2019, 20(7): 397-424.
[2] Coates M D, Tekin I, Vrana K E,.Review article: The many potential roles of intestinal serotonin (5-hydroxytryptamine, 5-HT) signalling in inflammatory bowel disease [J]., 2017, 46(6): 569-580.
[3] Walther D J, Bader M.A unique central tryptophan hydroxylase isoform [J]., 2003, 66(9): 1673-1680.
[4] del Colle A, Israelyan N, Gross Margolis K.Novel aspects of enteric serotonergic signaling in health and brain-gut disease [J]., 2020, 318(1): G130-G143.
[5] Scotton W J, Hill L J, Williams A C,.Serotonin syndrome: Pathophysiology, clinical features, management, and potential future directions [J]., 2019, 12: 1178646919873925.
[6] Zhang X H, Perez-Sanchez H, Lightstone F C.A comprehensive docking and MM/GBSA rescoring study of ligand recognition upon binding antithrombin [J]., 2017, 17(14): 1631-1639.
[7] Kongsted J, Ryde U.An improved method to predict the entropy term with the MM/PBSA approach [J]., 2009, 23(2): 63-71.
[8] Ru J L, Li P, Wang J N,.TCMSP: A database of systems pharmacology for drug discovery from herbal medicines [J]., 2014, 6: 13.
[9] Camilleri M.LX-1031, a tryptophan 5-hydroxylase inhibitor, and its potential in chronic diarrhea associated with increased serotonin [J]., 2011, 23(3): 193-200.
[10] Lipinski C A, Lombardo F, Dominy B W,.Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings [J]., 2001, 46(1/2/3): 3-26.
[11] Veber D F, Johnson S R, Cheng H Y,.Molecular properties that influence the oral bioavailability of drug candidates [J]., 2002, 45(12): 2615-2623.
[12] Ouyang L, He G, Huang W,.Combined structure-based pharmacophore and 3D-QSAR studies on phenylalanine series compounds as TPH1inhibitors [J]., 2012, 13(5): 5348-5363.
[13] Zhong H, Huang W, He G,.Molecular dynamics simulation of tryptophan hydroxylase-1: Binding modes and free energy analysis to phenylalanine derivative inhibitors [J]., 2013, 14(5): 9947-9962.
Exploring potential TPH1 inhibitors from traditional Chinese medicine natural product database based on computer simulation technology
SHI Hai-long, CHENG Yi, HUANG Yue, FENG Xue-song, WANG Yue-wen, HUANG Feng, CHAO Xu
Shaanxi University of Chinese Medicine, Xi’an 712046, China
To discover the active molecules with inhibitory activity to tryptophan hydroxylase-1 (TPH1) from traditional Chinese medicine natural product database based on computer simulation technology.A drug screening platform, that integrates molecular docking, drug-like screening, pharmacokinetic prediction, and molecular dynamics simulation, was built to discovery TPH1 inhibitors from the TCMSP database.Chuanbeinone (TCMSP_ID: MOL009572) showed good inhibitory activity to TPH1 and drug-likeness, which can effectively inhibit gastrointestinal related TPH1 enzymes, but had weak inhibitory effect on central nervous system-related TPH2 enzyme activity.Through preliminary verification of enzyme activity inhibition, molecular dynamics simulation is used to accurately predict the free binding energy of THP1 and MOL009572 and various energy contributions.Chuanbeidone, MOL009572, an active molecule of traditional Chinese medicine that relieves irritable bowel syndrome-related pain, has been screened out through integrated multiple virtual screening technologies.
molecular docking; molecular dynamics simulation; tryptophan hydroxylase; inhibitor; chuanbeinone
R284.1
A
0253 - 2670(2022)10 - 2968 - 09
10.7501/j.issn.0253-2670.2022.10.005
2021-11-21
陕西省中医管理局中医药科研课题(JCMS003);陕西中医药大学创新团队项目(2019-Y505)
史海龙,男,博士,美国UNMC博士后,副教授,研究方向为计算机辅助药物设计及生物大分子模拟。E-mail: shl112@sntcm.edu.cn
通信作者:晁 旭,博士,教授,研究方向为中医药防治消化系统肿瘤。
[责任编辑 王文倩]
