Selective adsorption of propene over propane on Li-decorated poly(triazine imide)
2022-05-22YongWangXiaoxiaJiaLiboLiJiangfengYangJinpingLi
Yong Wang ,Xiaoxia Jia ,Libo Li ,Jiangfeng Yang Jinping Li
aCollege of Materials Science and Engineering,Taiyuan University of Technology,Taiyuan,030024,China
b Shanxi Key Laboratory of Gas Energy Efficient and Clean Utilization,Taiyuan University of Technology,Taiyuan,030024,China
c College of Chemistry and Chemical Engineering,Taiyuan University of Technology,Taiyuan,030024,China
Abstract Solid adsorbents that simultaneously have high selectivity and uptake capacity are highly promising as alternatives to conventional cryogenic distillation of propene/propane(C3H6/C3H8)separation.Coordinatively unsaturated metal sites(CUS)plays a vital role in selective adsorption of olefins over paraffins.Ultrathin poly (triazine imide) (PTI) nanosheets can reach rapid gas adsorption equilibrium,due to its large surface-tovolume ratio.In this work,combining the advantages of the CUS and the PTI nanosheets,Li CUSs were introduced into the PTI nanosheets for C3H6/C3H8 separation.Density functional theory(DFT)calculations demonstrated the thermodynamic feasibility of incorporating Li CUSs into the PTI nanosheets.These highly exposed Li CUSs were predicted to have a higher adsorption affinity toward C3H6 than C3H8.Using the DFTderived force field parameters,we further performed grand canonical Monte Carlo(GCMC)simulations to investigate C3H6/C3H8 adsorption on the Li-PTI complexes slit pore model with different pore widths (H).We found that the Li-PTI complexes display considerable C3H6/C3H8 selectivity (4.2-7.9) under relevant conditions.Moreover,the Li-PTI complexes slit pore have large C3H6 working capacities (1.5-4.0 mmol g-1),superior to those calculated for the most of adsorbent materials that have been reported.The Li-PTI complexes with slit pore architecture show potential as C3H6/C3H8 separation materials.
Keywords: Adsorption;Propene;Propane;Two-dimensional materials;Coordinatively unsaturated metal sites
1.Introduction
Propene,as an important commercial petrochemical,is in strong,constantly growing demand [1-3].The production of propene by cracking of fossil fuels yields mixture of propene and propane.To obtain high quality pure product,the separation of propene/propane (C3H6/C3H8) is a required process.Unfortunately,the industrially highly relevant separation of propene/propane belongs to the problematic cases,due to the similar molecular sizes and boiling points of these two molecules [4,5].Compared with current conventional,energyintensive,costly cryogenic distillation,separation methods based on adsorption such as vacuum or pressure swing adsorption (VSA or PSA) are desirable,as a result of their potential tremendous energy savings [6-11].The key for the adsorption processes is the adsorbent materials[12].However,the development of materials that simultaneously have high selectivity,large working capacity,fast adsorption kinetics and low production cost for C3H6/C3H8separation is a challenging task [13].In particular,some adsorbent materials used for the separation of olefins/paraffins exhibit high propene selectivity,but show poor olefins working capacities under relevant conditions [14].
Up to now,carbons,zeolites,and metal-organic frameworks(MOFs) have been widely reported as olefin adsorbents in the literatures [15-18].Among these olefin adsorbents,some materials containing coordinatively unsaturated metal sites(CUS)exhibit pronounced separation performance for olefins/paraffins due to the strong π-double-bond interactions between the CUS and olefins [19-23].Inspired by these findings,here we show another promising system:two-dimensional poly (triazine imide)(PTI)coordinated CUS.Recently,a variety of ultrathin graphene-like materials have been proposed for gas adsorption and separation [24,25].As a new family of graphitic carbon nitrides,PTI,with a graphene like framework,contains imidelinked triazine (C3N3) units [26,27].Experiments have demonstrated that the crystalline PTI can be cleaved into graphene-like nanosheets[26,28].Moreover,the rich electron lone pairs of pyridine-like nitrogen in PTI may serve as chelate bonding sites to capture metal atom[29-32].Based on this fact,we demonstrated that the CUS could be introduced in the PTI nanosheets.The complexes of CUS and PTI nanosheets have three advantages over the widely reported C3H6/C3H8separation materials modified with CUS:(i)compared with the single coordination site that the limited metal ions expose in MOFs,more exposed CUSs could be found in the PTI nanosheets.They may be expected to exhibit potentially even better propene/propane separation performance.(ii) the ultrathin PTI nanosheets have large surface-to-volume ratio,and can reach rapid gas adsorption equilibrium [33,34].(iii) the low-cost raw materials and high stability of the PTI nanosheets.Thus,by the density functional theory(DFT)calculations and grand canonical ensemble Monte Carlo (GCMC) simulations,we investigated the C3H6/C3H8adsorption properties of the PTI nanosheets coordinated metal ions.Here,we selected Li-PTI complexes as a representative model of M-PTI nanosheets since Li with light weight and low cohesive energy would be more suitable choices of the metal ions that inserted into the PTI nanosheets,compared with heavier metal ions[35].
2.Models and methods
2.1.Density functional theory calculations
As shown in Fig.1(a),the geometry of monolayer PTI adopted in the computational study was taken from the experimental PTI crystal structure [26].With full cell optimization and all atoms free to move,the geometry optimizations of monolayer PTI and its complexes were performed by dispersion-corrected DFT (DFT-D2) calculations in DMol3package implemented in the Materials Studio software[36,37].A vacuum region of 20 Å in the z-direction and a 2 × 2 supercell were used.Perdew-Burke-Ernzerhof (PBE)functional under the generalized gradient approximation(GGA) and spin-unrestricted method were adopted [38].A double numerical plus polarization basis set was employed in all the calculations and core electrons were used to set the type of core treatment [39].The value of real-space global cutoff radius was 6.0 Å.10-6Ha (energy),5 × 10-4Ha Å-1(gradient),and 5 × 10-3Å (displacement) were used as the convergence threshold optimization parameters.A k-point separation of~0.01 Å-1in the reciprocal space at the periodic directions obtained fromK-point meshes of 7 × 7 × 1 was used [40].
2.2.Grand canonical ensemble Monte Carlo (GCMC)simulations
C3H6and C3H8were modeled using the united atom model,where the CHxgroups were treated as a single interaction center with effective potential parameters.For C3H6,we used a point charge model (Propene PC-Lgg) from Gutiˊerrez-Sevillano et al.[41]The TraPPE force field was used for modeling C3H8[42].The angles and bond lengths of the gas molecules were considered to be rigid.The Li-PTI complexes were modeled as rigid frameworks.Except those for the Li atoms in the framework,the LJ potential parameters from the DREIDING force field for the framework were adopted [43].The LJ potential parameters for Li atoms were taken from the UFF force field[44].For the atomic partial charges of the Li-PTI complexes,DFT-derived Mulliken charges were used.

Fig.1.Geometric configurations of (a) PTI nanosheets and (b) Li-PTI complexes after geometry optimisation (C,gray;N,blue;H,white;and Li,purple).
All GCMC simulations were c arried out with Sorption module in the Material Studio software [45].For gas adsorption simulations on the surface of the Li-PTI complexes,the simple slit pore model,as a representative carbon pore architecture,was adopted in this study.Using two Li-PTI nanosheets of 3.5 × 3.5 nm,we built the slit-pore model(Fig.S3).The pore width (H) measured by the distance between the two Li-PTI nanosheets was varied as 0.8,1.2,1.6,and 2.0 nm.For all three dimensions,periodic boundary conditions were used.The pressure was converted into the corresponding fugacity by using the Peng-Robinson equation of state.2 × 107moves were used for each equilibration and 2 × 107moves were used for calculating ensemble averages[46].The intermolecular interactions were calculated with the site-site Lennard-Jones (LJ) and shielded potentials.The cutoff radius was set to 12 Å for the LJ interactions.The Ewald summation technique was used to describe the longrange electrostatic interactions.
3.Results and discussion
3.1.Structural properties
Fig.1(b) shows the optimized structure of Li-PTI complexes.It is observed that the Li atom is connected to two adjacent pyridinic-N atoms forming a six-member ring.The length of Li-N bonds is 0.1994 nm,which is comparable to the value of 0.1990 nm obtained from Li-N bonds in MOFs[47].The binding energy between Li and PTI nanosheets is-216.8 kJ mol-1,which is stronger than the cohesive energy of bulk Li (-157.3 kJ mol-1),avoiding clustering of the Li atoms [48].Li-PTI complexes are stable structure with dispersive Li atoms on the PTI nanosheets.As a comparing,the binding energy between Li and graphene is only-106.1 kJ mol-1,smaller than that of bulk Li[49].To further examine the type of Li-N bonds,we also calculated the Mulliken charge and the PDOS of Li-PTI complexes.The Mulliken charge calculation result shows that Li atoms transfer excessive charge (0.154 e-) to the N atoms and become positively charged.In this case,the Li shows strong static Coulomb interaction with the PTI nanosheets.Lee et al.[50] have reported that the lowest unoccupied molecular orbitals of Li atoms can be hybridized with the highest occupied molecular orbitals of adsorbents,resulting in bonding and antibonding formation.Fig.2(a)shows the partial density of states(PDOS).One can see peaks overlaps between the Li 2p orbitals and N 2p orbitals,indicating the bond formation between the Li and the PTI nanosheets.Therefore,the polarization and the hybridization mechanisms both result in the adsorption of Li atoms on the PTI nanosheets.It should be noted that Li,together with organic linkers,can form MOFs in which Li is four-coordinated[51].In this study,the Li use two coordination sites to form Li-N bonds with two pyridinic-N atoms by bonding in the PTI nanosheets.The other two coordination sites are unoccupied.The results present that the coordination sites of PTI nanosheets can capture Li atoms to form highly exposed CUS in the PTI nanosheets.
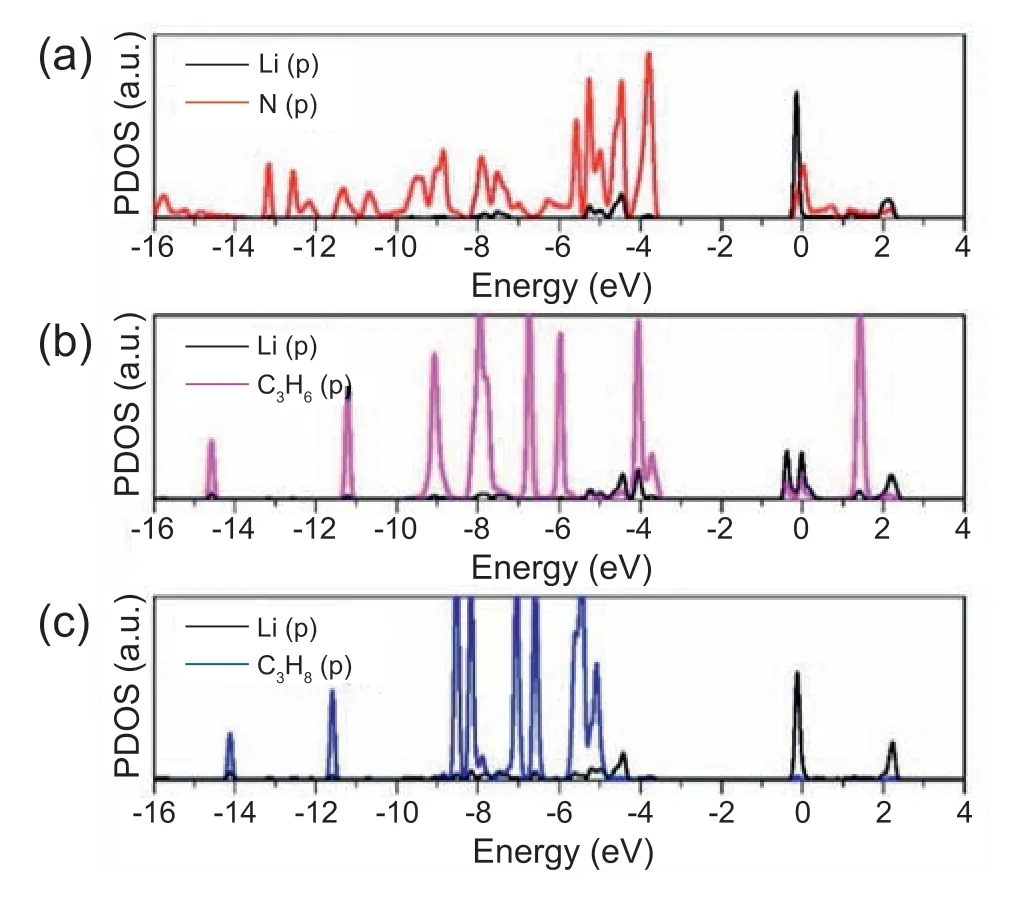
Fig.2.PDOS of (a) Li atom and N atom nearby the Li atom in the Li-PTI complexes,(b) Li atom and C3H6 molecule for the C3H6 adsorbed Li-PTI complexes,and (c) Li atom and C3H8 molecule for the C3H8 adsorbed Li-PTI complexes.
3.2.Gas adsorption on the Li-PTI complexes
Next,the C3H6/C3H8adsorpton properties of the Li-PTI complexes surface were considered.The most stable adsorption configurations of C3H6and C3H8on the Li-PTI complexes surface are shown in Fig.3.It can be seen that the C3H6molecule is preferred adsorbed on the Li CUS due to the strong π-complexation between C3H6and Li CUS.From Fig.2(b),we can observe peaks overlaps around -4 eV as consequence of the hybridization between the empty Li's p orbital and C3H6's π-orbital.While the peaks overlaps around 0 eV could be attributed to the electron backdonation from the p orbital of Li site to the π*antibonding orbital of C3H6.The bonding and backbonding synergistically contribute to the adsorption of C3H6on the Li-PTI complexes surface,with binding energy of -54.5 kJ mol-1.The average distance between Li site and the two C=C carbon atoms of C3H6is 0.2390 nm.C3H8is adsorbed on the above of the three Li atoms with one CH3group is pointing to one Li atom with a distance of 0.2464 nm.The overall PDOS change of Li atoms due to the C3H8adsorption is negligible(Fig.2(c)),indicating that the interactions between C3H8and Li-PTI complexes surface are based on physical adsorption.The binding energy of C3H8is found to be -30.1 kJ mol-1.The difference in binding energy between the adsorbed C3H6and C3H8on the Li-PTI complexes surface is as high as 24.2 kJ mol-1.One can expect a higher C3H6/C3H8selectivity on the Li-PTI complexes surface.

Fig.3.Most stable state for a single(a)C3H6 molecule and(b)C3H8 molecule absorbed on the Li-PTI complexes surface (C,gray;N,blue;H,white;Li,purple;C3H6,green;and C3H8,orange).
Motivated by the preferential binding of C3H6by the Li-PTI complexes surface,GCMC simulations for the adsorption behaviours of C3H6and C3H8on the Li-PTI complexes surface were further performed.As discussed previously,a quantum chemical treatment is needed to model the strong coordination-like interactions between the CUS of Li and the double bond of C3H6.The strong coordination-like interactions are hard to model using generic force field [52,53].Thus,the force field parameters derived from DFT calculations were introduced to describe the interactions between C3H6and Li CUS of the Li-PTI complexes surface.Firstly,based on the most stable state of C3H6on the Li-PTI complexes surface,we obtained the interaction energy profiles for C3H6with changing the distance between the C=C bond of the adsorbed C3H6molecule and Li CUS.At each different C3H6-Li distance,the interaction energy was computed by DFT-D2 and classical molecular mechanics calculations(Forcite module in the Material Studio software)from Eq.(1):

whereEpropene-Li-PTIComplexis the total energy of the propene and Li-PTI complex,ELi-PTIComplexis the energy of the isolated Li-PTI complexes andEpropeneis the energy of C3H6molecule.From Fig.4,one can observe that the interaction energies are much lower in the case of the classical molecular mechanics calculations with generic force field than that obtained from the DFT-D2 calculations.Thus,a dummy interaction site was added between the two C=C carbon atoms in the C3H6,which interacts only with the Li CUS and denoted by Li-π vdW interaction site.Jorge and co-workers have successfully used this method to investigate the gas adsorption performance of MOF materials [54,55].The Li-π vdW interaction energy (Red squares in Fig.4) can be computed from DFT-D2 interaction energy subtracting the generic force field contributions.We fitted Li-π vdW interaction to a Buckingham potential:


Fig.4.The interaction energies of C3H6 on the different distances between the center of the C=C bond of C3H6 molecule and the closest Li atom in the Li-PTI complexes.Black circles and blue triangles are data obtained from the DFT-D2 calculations and the classical molecular mechanics calculations with generic force field,respectively.Red squares are data obtained from the difference between DFT-D2 interaction energy and the generic force field contribution,while the full red line is the fitting of Eq.(2).The full black line is obtained by the addition of the Li-π vdW fit to the generic force field contributions.
whereAis the exponential coefficient,β is the Buckingham β parameter,C6is the Buckingham C6 parameter,Ris the distance from the Li CUS to the center of the C=C bond in the C3H6molecule.Then the obtained potential energy function of Li-π vdW interaction (Table S4) was added to the generic force field.As shown in Fig.4,the fit is very good and the different between DFT-derived data and the developed force field is very small.Additionally,the correct trend of C3H8uptakes was obtained through DFT calculation of Li-C3H8(interaction between C3H8and the Li-PTI complexes surface),followed by fitting of Morse force field parameters.The details are shown in Supporting Information (SI).Finally,we further refitted the above potential parameters by multiplying a constant value to reduce the difference between the gas binding energy obtained from DFT-D2 calculations and the isosteric heats of gas adsorption(Q0st)near the zero-coverage obtained from GCMC simulations at 0 K.The factors for C3H6and C3H8adsorption are 0.58 and 0.83,respectively.As a validation of the DFT-derived force field,the isosteric heat of adsorption (Qst) and adsorption isotherms for pure C3H6and C3H8on the one side of monolayer Li-PTI complexes at 298 K were calculated.As shown in Fig.S5,theQstvalues at lower gas loading for C3H6and C3H8are-48.8 kJ mol-1and-22.6 kJ mol-1,respectively.A noteworthy point is that the binding energy might be reduced by the neglect of zero-point energy and other thermal correction by about 5 kJ mol-1[56,57].Thus,our results using the DFT-derived force field are consistent with the binding energies of C3H6(-54.5 kJ mol-1) and C3H8(-30.1 kJ mol-1) obtained from DFT calculations.
Using the DFT-derived force field,we obtain equimolar C3H6/C3H8adsorption isotherms at 298 K and 10-3-1 bar.Since GCMC simulation results only provide the absolute number of guest molecules in the framework (absolute loadings,Nabs) [58],the excess gas uptake were calculated using the Eq.(3):

whereNexandNabsrepresent the excess loading and absolute loading,respectively.ρgis the gas density obtained from Peng-Robinson formulation with constant temperature and pressure.Vgis the free volume.Fig.5(a) and (b) show the calculated results.An abrupt isotherm of C3H6increase at low pressures is observed,corresponding to the strong interaction of C3H6with the Li-PTI complexes surface.The C3H6uptake is much higher than C3H8uptake,indicating that the investigated Li-PTI complexes selectively adsorb C3H6over C3H8.
In addition,the pore widths show a large influence on the gas adsorption,which can result from the strong overlap interaction from Li-PTI nanosheets or the gas adsorption layers whenHis small.At 298 K and 1bar,the C3H6and C3H8absorbed amounts of Li-PTI complexes (H=1.2 nm) can be up to 1.98 and 8.62 mmol g-1,respectively,which are the highest among the Li-PTI complexes.Considering that the small simulation boxes (3.5 × 3.5 nm) might affect the statistics of GCMC simulation,a larger model was constructed with dimensions of about 7×7 nm to check the suitability of the model used in our simulation.As shown in Fig.S6,the model size of Li-PTI complexes (H=1.2 nm) has almost no influence on the GCMC simulation results.With decreasing temperature,the C3H6and C3H8unptake capacity on the Li-PTI complexes increase considerably at 273 K (Fig.S8).It is notable that Li-PTI complexes(H=1.2 nm)exhibit a very pronounced S-shaped isotherms.With optimizing pore size,the attractive electrostatic interactions between gas molecules are responsible for the unusual shape of the adsorption isotherms [59].

The adsorption selectivity is given by wherexiandyiare the mole fractions of component i(i=1,2)in the adsorbed and bulk phases,respectively.In this study,component 1 and component 2 are C3H6and C3H8,respectively.The adsorption selectivity for the equimolar C3H6/C3H8mixture at 298 K are shown in Fig.5(c).The C3H6/C3H8selectivity decrease as pressure increase.At 1 bar,the adsorption selectivity obtained for the Li-PTI complexes decrease to 4.4-7.7 depending on the pore width,which are still considerable.The working capacity is another key evaluation criterion for comparing the separation performance of various adsorbents in the PSA or VSA process [46].The high selectivity result in an enhanced purification efficiency of C3H6,while the large working capacity means the lower overall cost.In this study,the C3H6working capacity of the Li-PTI complexes is defined as the difference between the C3H6amounts in the adsorbed gas mixture at the adsorption pressure(1 bar)and the desorption pressure(0.1 bar).Fig.5(d)and Table S6 show the comparison of the C3H6working capacities and C3H6/C3H8selectivity for selected adsorbent materials reported to date.Although the C3H6/C3H8selectivity of the Li-PTI complexes with slit pore architecture are lower than those of some adsorbent materials,the obtained C3H6working capacities are considerably large.Especially for the Li-PTI complexes(H=1.2 nm),the C3H6working capacity is 4.0 mmol g-1,which is rare in an adsorbent material.It should be emphasized that for most practical applications,a high working capacity is more important than a high selectivity.Therefore,the Li-PTI complexes are promising sorbent for C3H6/C3H8adsorption separation.

Fig.5.Adsorption isotherms of(a)C3H6 and(b)C3H8,(c)the simulated C3H6/C3H8 selectivity in equimolar C3H6/C3H8 mixture for the Li-PTI complexes with different pore widths at 298 K.(d)C3H6 working capacities as a function of C3H6/C3H8 selectivities for the selected adsorbent materials reported to date under an equimolar gas mixture.
4.Conclusions
By considering the combination of Li CUS and PTI nanosheets,the C3H6/C3H8adsorptions on this Li-PTI complexes surface were investigated.The binding energy of Li on PTI nanosheets is shown to be much greater than the cohesive energy of bulk Li.Hence the Li CUSs can distribute on PTI nanosheets without any cluster formation.Mulliken charge and PDOS analysis illustrate that the high binding energy of Li on the PTI nanosheets is attributed to the polarization and hybridization mechanisms.Due to the significant contribution of π complexation interaction between C3H6and Li CUS,an increase of 24.2 kJ mol-1was observed when compared to C3H8.The significantly stronger binding energy of Li-PTI to C3H6suggests that the Li-PTI complexes may be useful for separating C3H6/C3H8mixture.The subsequent GCMC simulations with DFT-derived field further confirm that the Li-PTI complexes with slit pore architecture simultaneously provide the considerable C3H6/C3H8selectivity and significantly large C3H6working capacity.These results could provide some useful information to active experiments in developing two-dimensional ultrathin materials for adsorption-based C3H6/C3H8separation.
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos.21706180).This research used resources of the Taiyuan University of Technology Quantum Chemistry Research Group.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gee.2020.10.001.
杂志排行

Green Energy & Environment的其它文章
- Investigating ionic liquids for optimizing lithium metal anode
- A mini-review on ZnIn2S4-Based photocatalysts for energy and environmental application
- Advanced silicon nanostructures derived from natural silicate minerals for energy storage and conversion
- B-doped activated carbon as a support for a high-performance Zn-based catalyst in acetylene acetoxylation
- High recycling Fe3O4-CdTe nanocomposites for the detection of organophosphorothioate pesticide chlorpyrifos
- Hierarchical Cu3P-based nanoarrays on nickel foam as efficient electrocatalysts for overall water splitting