PYCR1基因突变致遗传性皮肤松弛症IIB型1例并文献复习
2022-05-22许照洁曹攀龙陈淑芳王春林
许照洁 曹攀龙 陈淑芳 王春林
患儿女,3岁,因“身材矮小”于2020年9月22日就诊于浙江大学医学院附属第一医院儿科门诊。患儿孕40周出生,出生体质量1 450 g,出生身长45 cm,运动、语言发育落后,8个月会坐,2岁会走。家族中无类似病史,父母体健,非近亲结婚。体格检查(图 1):身高 88 cm(-2 SDS),体重 12 kg(-1.4 SDS),神志清,皮肤松弛,静脉显露,特殊面容(倒三角脸,前额突出,面中部发育不全,发际线高,招风耳,睑裂下斜),心肺腹无殊,手指细长,拇指内收,关节松弛,乳晕无色素沉着,双乳B1期,阴蒂肥大,阴毛PH1期。辅助检查:肝肾脂糖电解质、抗缪勒氏管激素、皮质醇、促肾上腺皮质激素、脱氢表雄酮、雄烯二酮、17α羟孕酮、血清胰岛素、糖化血红蛋白未见异常;染色体46,XX。
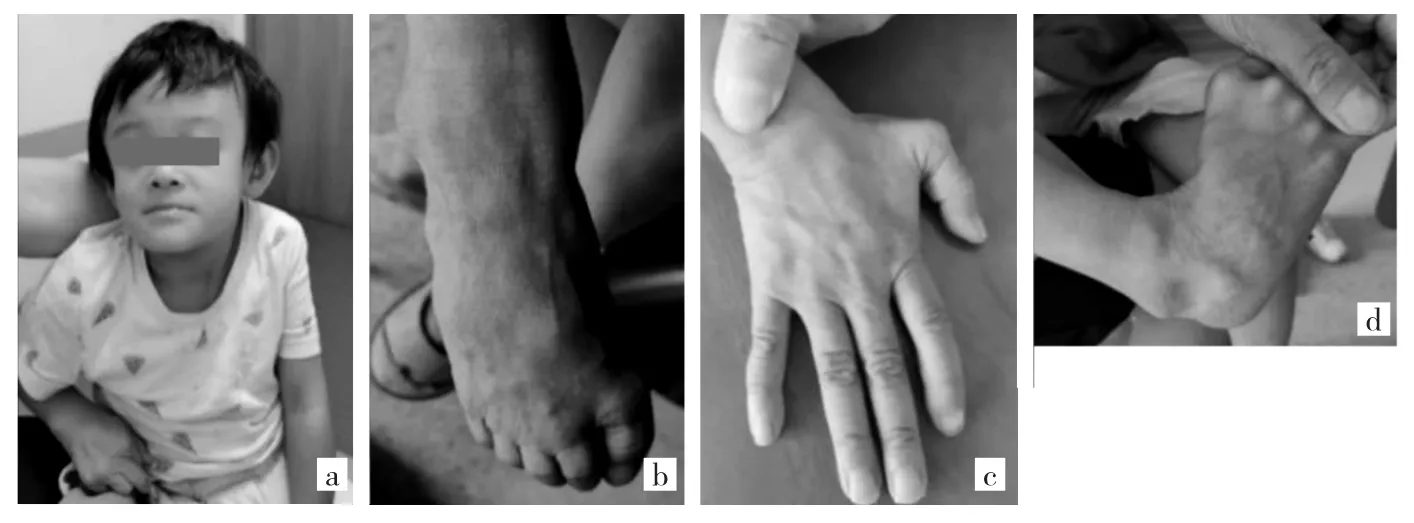
图1 患儿面部、皮肤及关节特征(a:倒三角脸,前额宽大而突出,招风耳;b:皮肤皱纹、松弛,静脉显露;c:皮肤皱纹,拇指内收;d:关节过度松弛)
经患儿家长知情同意后进行基因检测,trio全外显子组测序采用IDT The xGen Exome Research Panel v2.0全外显子捕获芯片(美国IntegratedDNATechnologies公司),经Illumina NovaSeq 6000系列测序仪(PE150,美国 Illumina公司)测序完成。全外显子测序发现患儿PYCR1基因 存在 2个变异(图 2):c.751C>T(p.Arg251Cys)、c.743G>A(p.Gly248Glu),经Sanger测序验证,其父亲携带c.751C>T突变,其母亲携带c.743G>A突变。该变异先证者为杂合子,符合常染色体隐性遗传复合杂合遗传疾病发病机制。结合患儿临床表现,诊断为常染色体隐性遗传性皮肤松弛症ⅡB型(autosomal recessive cutis laxa type 2B,ARCL2B)。既往曾有过 c.743G>A突变的文献报道[1],但c.751C>T为新发现的变异位点,扩大了PYCR1基因谱。

图2 患儿及父母基因测序图(患儿PYCR1基因存在c.751C>T(p.Arg251Cys)和c.743G>A(p.Gly248Glu)复合杂合突变,分别来源于父亲和母亲)
PYCR1基因位于17号染色体(17q 25.3),编码存在于线粒体中的吡咯啉-5-羧酸还原酶1。PYCR1酶是多聚体蛋白复合体,由5个相同的多肽亚基形成五聚体环状结构(图3a-b),再由两个相互作用的五聚体通过C-末端结构域构建为十聚体复合物,具有生物活性[2]。图3c为PYCR1蛋白在两个变异位点的野生型模型,图3 d为软件预测该患儿PYCR1蛋白p.Arg251Cys和p.Gly248Glu局部变体结构。讨论 皮肤松弛症是一组以皮肤松弛为主要表现,伴或不伴内脏器官受累的罕见遗传性或获得性结缔组织疾病,涉及多个系统,包括骨骼、心血管、呼吸及神经等。遗传性皮肤松弛症根据遗传方式分为5型:常染色体显性遗传、伴性遗传、常染色体隐性遗传Ⅰ型、常染色体隐性遗传Ⅱ型、常染色体隐性遗传Ⅲ型。该患儿为由PYCR1基因突变引起的 ARCL2B(MIM 612940)。PYCR1基因编码的吡咯啉-5-羧酸还原酶1催化吡咯啉5-羧酸盐转化为脯氨酸,脯氨酸的降低会影响结缔组织中胶原和弹性蛋白的合成,导致皮肤松弛[3]。Huang等[4]研究发现,与健康者比较,PYCR1突变患者的皮肤成纤维细胞中膜蛋白、弹性蛋白、核心蛋白、五角蛋白3等基因表达明显降低。PYCR1酶在骨骼和皮肤中表达最高,除皮肤表现之外,PYCR1突变还会导致骨关节异常、生长发育异常等。

图3 PYCR1酶及变异蛋白空间结构(a:五聚体蛋白复合体;b:其中1个亚基;c:PYCR1野生型;d:PYCR1蛋白p.Arg251Cys,p.Gly248Glu局部变体结构)
检索中英文数据库,筛选出有基因结果及临床资料的PYCR1基因突变引起ARCL2B 的文献共 16 篇[1,5-19],报道了 107例病例,加上本例共108例。统计ARCL2B的主要临床表现及出现频率如下:皮肤松弛及皱纹(100.0%)、关节松弛(98.9%)、特殊面容(97.1%)、宫内发育迟缓(94.4%)、精神运动发育迟缓(94.3%)、皮肤变薄或静脉显露(85.5%)、骨质疏松(82.4%)、前囟过大或闭合延迟(82.4%)、生长发育不良(79.2%)、张力减退(66.0%)、手指挛缩或拇指内收(65.3%)、小头畸形(62.3%)、髋关节脱位(58.1%)、胼胝体发育不全(48.1%)、疝气(38.5%)、蓝巩膜(34.4%)、斜视(31.7%)、手足徐动(21.1%)、白内障(14.0%)。部分特异性表型如眼部异常(蓝巩膜、斜视、白内障)、胼胝体发育不全、手足徐动等,具有疾病预测价值。
在报道的病例中,约85%的突变位于外显子4、5和6,常见的突变位点如c.540+1G>A[6,9,11,17]15 例(13.9%)、c.797G>A[7,9,13-14]10 例(9.3%)、c.797+2_797+5del[6,9]9 例(8.3%)、c.616G>A[6,9,16,19]6 例(5.6%)等,约12%为复合杂合突变。错义突变是最常见的突变类型(48例,44.4%),其次是剪接位点突变(38例,35.5%),另外几种突变类型所占比例均不高,移码突变7例(6.5%),剪接位点突变和错义突变的组合6例(5.6%),移码突变和错义突变的组合2例(1.9%),无义突变4例(3.7%),PYCR1全基因缺失3例(2.8%)。将患者根据其突变类型分成7组后进行Fisher确切概率法检验,除疝(P<0.01)及精神运动发育迟缓(P<0.01)外,其余不同突变类型之间临床表型比较差异均无统计学意义(均P>0.05)。进一步进行组间两两比较后发现,只有对于精神运动发育迟缓的发生频率,PYCR1全基因缺失组与其他各组之间差异均有统计学意义(均P<0.05)。Dimopoulou 等[6]发现,与外显子4、5和6的突变相比,临床特征不明显以及没有或仅轻度智力残疾的患者与外显子1和2的突变有关,与此相似,本文也发现精神运动发育迟缓的发生与突变类型也有关,共有6例患者无精神运动发育迟缓表现,其中包括2例c.138+1G>A纯合子突变[6,9]和 3例 PYCR1全基因缺失[16]。有一个假设可以解释此现象,PYCR1是一种线粒体内蛋白,突变位于外显子1和2可能导致无义介导的mRNA降解,和PYCR1全基因缺失一样,导致PYCR1蛋白完全缺失,而且位于1、2外显子的错义突变,可能会改变N端的靶信号从而影响线粒体导入和亚线粒体定位。线粒体中蛋白的减少,无论是由于mRNA或基因产物的完全缺失还是由于缺陷蛋白导入,对组织功能的危害要小于因蛋白的某种改变而不能形成同源十聚体并执行其酶功能的危害[6]。
ARCL2B需与多种疾病鉴别。轻度患者与骨发育异常性皮肤老化症(geroderma osteodysplasticum,GO)的表型相似,GO 通常智力发育正常[11],特征包括手足背部的皮肤松弛及皱纹、下颌发育不全、骨质疏松、病理性骨折、长骨弯曲等[14]。皱纹皮肤综合征(wrinkly skin syndrome,WSS)也可表现为皮松皱纹及松弛、生长发育迟缓、小头畸形、关节脱位等,但WSS的皮肤变化及面部畸形较ARCL2B轻,常常仅限于面中部发育不全[20]。另外,皮肤松弛症各型均有皮肤松弛表现,尤其ARCL2B与常染色体隐性遗传Ⅲ型(又名为德巴西综合征,De Barsy syndrome,DBS)表型高度相似,DBS也可表现为早老现象、骨骼及眼部异常等,可能智力残疾更为严重[21],还有报道过先天性青光眼、主动脉根部扩张和先天性肥厚型幽门狭窄这些特殊表现[22]。很多PYCR1突变的皮肤松弛患者,曾被诊断为WSS、GO、DBS,基因诊断后重新被分类至ARCL2B[7,9],提示需要进一步的基因检测才能更准确地区分和诊断这类疾病。
总的来说,ARCL2B临床表现多样,与多种疾病有相似的临床特征,如在临床碰到有皮松皱纹、特殊面容、关节松弛、骨质疏松、宫内发育迟缓、身材矮小、精神运动发育迟缓等皮肤、关节、骨骼及生长发育异常的患儿,需考虑PYCR1基因相关ARCL2B,并行基因检测进一步明确诊断。
