基于血清miRNA相对表达秩序关系识别乳腺癌预警标志
2022-05-20赵文倩龙永金刘玉婷尹博杰
赵文倩,曾 婷,郭 俊,龙永金,刘玉婷,张 勤,尹博杰,李 娜
(1.赣南医学院2018级生物信息学专业本科生;2.赣南医学院医学信息工程学院,江西 赣州 341000)
乳腺癌是目前女性健康最具威胁的疾病之一[1],位于女性肿瘤发病率的首位,死亡率的第二位[2-3]。乳腺癌早期患者治愈率比较高,达到80%~90%[4],但早期患者病症并不明显,容易被忽视[5],因此需要一种敏感的、非侵入性的分子标志物来提高乳腺癌的早期检测准确率。血液是人体一种特殊的组织,具有采样容易、对身体的侵袭性小并易于临床实践和验证的优点,且与各组织器官之间都存在着交互作用[6-7]。血液中可能存在着与癌症相关的分子改变特征[8],这些分子改变特征可在血液或组织中被发现[9-10]。
microRNA(miRNA)是短的非编码RNA,长度约18~25 个核苷酸。miRNA 通过结合3'-UTR 的种子区下调靶基因mRNA在胞质内的表达,并且可以负向介导其丰度和翻译潜能[11]。miRNA 在多种生物过程包括癌症在内的各种疾病中发挥着重要作用[12-13]。越来越多的研究将miRNA作为癌症生物标志物[14-17],基于取样容易、侵袭性小的特点,在血清样本中寻找肿瘤相关的miRNA 风险标志成为研究热点。大量研究将血清中的miRNA 作为乳腺癌的诊断标志,例如利用135 例乳腺癌样本和125 例健康对照样本筛选得到的3-miRNA 标志[18],利用RT-qPCR检测miRNA的表达水平筛选得到的miR-24和miR-103a 标志[19],利用RT-qPCR 技术对30 例乳腺癌样本和20 例健康对照样本的miRNA-155 血清表达水平进行测量分析[20]等。研究者大多采用Logistics 回归、支持向量机、神经网络等机器学习算法对构建的标志计算风险打分,以此作为分类依据。但目前广泛采用的RNA-seq 测序技术、基因芯片技术和PCR技术并不能准确测量出绝对的miRNA表达水平,不同实验的检测结果往往存在较大差异,同一组风险标记在不同训练集中存在不同的风险打分,跨数据集能力差。因此,如何在血清样本中寻找稳定的具有临床转化价值的风险标记成为亟待解决的问题。本文拟在基于样本内miRNA 相对表达秩序关系,找出在血清中发生改变的miRNA作为筛选早期乳腺癌的预警标志。
1 材料和方法
1.1 数据本文所分析的血清miRNA 数据均来自GEO(Gene Expression Omnibus)数据库(https://www.ncbi.nlm.nih.gov/geo/),包含4套数据共计7 127例样本,见表1。所有数据集均由3D-Gene Human miRNA 检测平台进行检测,检测miRNA 数为2 526。这些数据均来自经预处理的series 数据集,数据预处理方式是对照组miRNA 按信号强度排列,去除前5%和后5%的表达值后计算平均值(M)和标准差(SD)。如果miRNA 的信号值大于M与2 倍的SD之和,则用miRNA 的信号值减去M作为该miRNA 的信号值。未检测到的miRNA 信号则用芯片上信号强度最小值减去0.1之后进行log2转换,然后进行分位数归一化。选择数据量最大的GSE73002 数据集作为训练集,包含2 686 例正常样本和1 280 例乳腺癌样本,其余3套数据集作为独立验证集。

表1 数据集的样本情况
1.2 筛选miRNA 标志对在训练集中基于样本内miRNA 相对表达秩序关系筛选稳定对,稳定对是指在正常对照样本中保持一致秩序关系的miRNAmiRNA。假设有两个miRNA,用i和j表示,表达水平用EmiRi和EmiRj表示,通过公式(1)计算miRNA 对(i,j)表达水平的秩序关系在正常样本中保持一致的概率P(EmiRi>EmiRj),如果概率满足P(EmiRi>EmiRj)≥0.95,则这个miRNA 对(i,j)定义为高度稳定对。公式(1)如下所示:

这里n表示正常样本数目,t表示第t个样本。
根据稳定对的计算公式,获得在95%的正常样本中秩序关系保持一致的高度稳定miRNA 对。在乳腺癌样本中,高度稳定miRNA 对秩序关系发生显著逆转的miRNA 对称为具有潜在预测能力的候选miRNA 标志对。利用卡方检验评价miRNA 对在两类样本中秩序关系的差异程度,显著性水平定义为P<0.01。
1.3 预警标志的分类模型基于多数投票规则,对待测样本进行判别。如果该样本检测的miRNA对中有半数及以上(即若标志对的对数N为奇数,则取N/2 的向上取整数对,若N为偶数则取(N/2 + 1)对)发生了逆转则判别为高风险样本,否则为低风险样本。公式⑵如下,f(x)表示发生逆转的miRNA对数,N为预警标志的对数。

1.4 miRNA靶基因及功能富集分析利用mirDIP靶基因整合数据库(http://ophid.utoronto.ca/mirDIP/index.jsp)寻找miRNA 的靶向基因。mirDIP 数据库对于来自多个数据库中的靶基因信息进行了归一化处理。将所有靶基因按照可信度从高到低排列,将其归一化到0~1 这个区间,0 代表可信度最高,1代表可信度最差,然后利用各个数据库中归一化之后的数值,给出一个总的打分值S(Score),公式⑶如下:

对获得的靶向关系构建miRNA-靶基因共表达网络。根据所获得的靶基因,利用注释、可视化和集成发现数据库DAVID 线上工具(Database for Annotation,Visualization and Integrated Discovery,DAVID v6.8,ncifcrf.gov)进行功能富集分析。
2 结 果
2.1 预警标志的构建基于样本内miRNA 相对表达秩序关系识别乳腺癌标志的主要流程如图1所示。

图1 识别乳腺癌miRNA预警标志流程图
利用训练集GSE73002 的1 280 例乳腺癌样本和2 686 例正常对照样本数据,基于样本内miRNA相对表达秩序关系,发现204 533 对在95%以上正常样本中秩序关系保持一致的高度稳定miRNA 对,其中122对在乳腺癌样本中保持10%以下的秩序关系,即在至少90%的乳腺癌样本中秩序关系发生显著逆转(P<0.01)。将122 对高度稳定对按照在乳腺癌样本中秩序关系比例进行升序排列,选择前11对miRNA-miRNA 作为预警标志(涉及13 个miRNA),如表2。图2显示11 对miRNA 和13 个miRNA 在训练集GSE73002 正常样本和乳腺癌样本中的表达情况,图2A 显示11 对miRNA 在两类样本中的表达具有明显差异,图2B 也显示13 个miRNA 在两类样本中具有表达差异性。该结果表明,基于样本内相对表达秩序关系的方法能在血清样本中筛选出特异性的分子改变特征。

表2 乳腺癌miRNA预警标志

图2 预警标志在正常样本和乳腺癌样本中的表达
2.2 预警标志的验证使用训练集和3 套独立验证集对预警标志进行检验。根据多数投票规则,对各数据集进行判别。各数据集的判别结果如表3所示,在乳腺癌样本和正常样本中,预警标志对样本的判别准确率为93%~100%,表明11对miRNA 可有效判别出乳腺癌患者。图3用受试者工作特征曲线(Receiver Operating characteristic Curve,ROC)[21]展示4 套数据的样本判别情况,曲线下面积AUC(Area Under Curve)反映了分类模型的好坏。图3A~3C 展示数据集GSE73002(AUC:0.970),GSE113486(AUC:1.000)和GSE106817(AUC:0.964)的ROC曲线。图3D展示样本判别正确的占比(具体数值见表3)。1 套训练集和2 套验证集的结果表明,基于样本内相对表达秩序关系构建的预警标志具有良好的稳健性。

表3 乳腺癌miRNA预警标志的判别结果
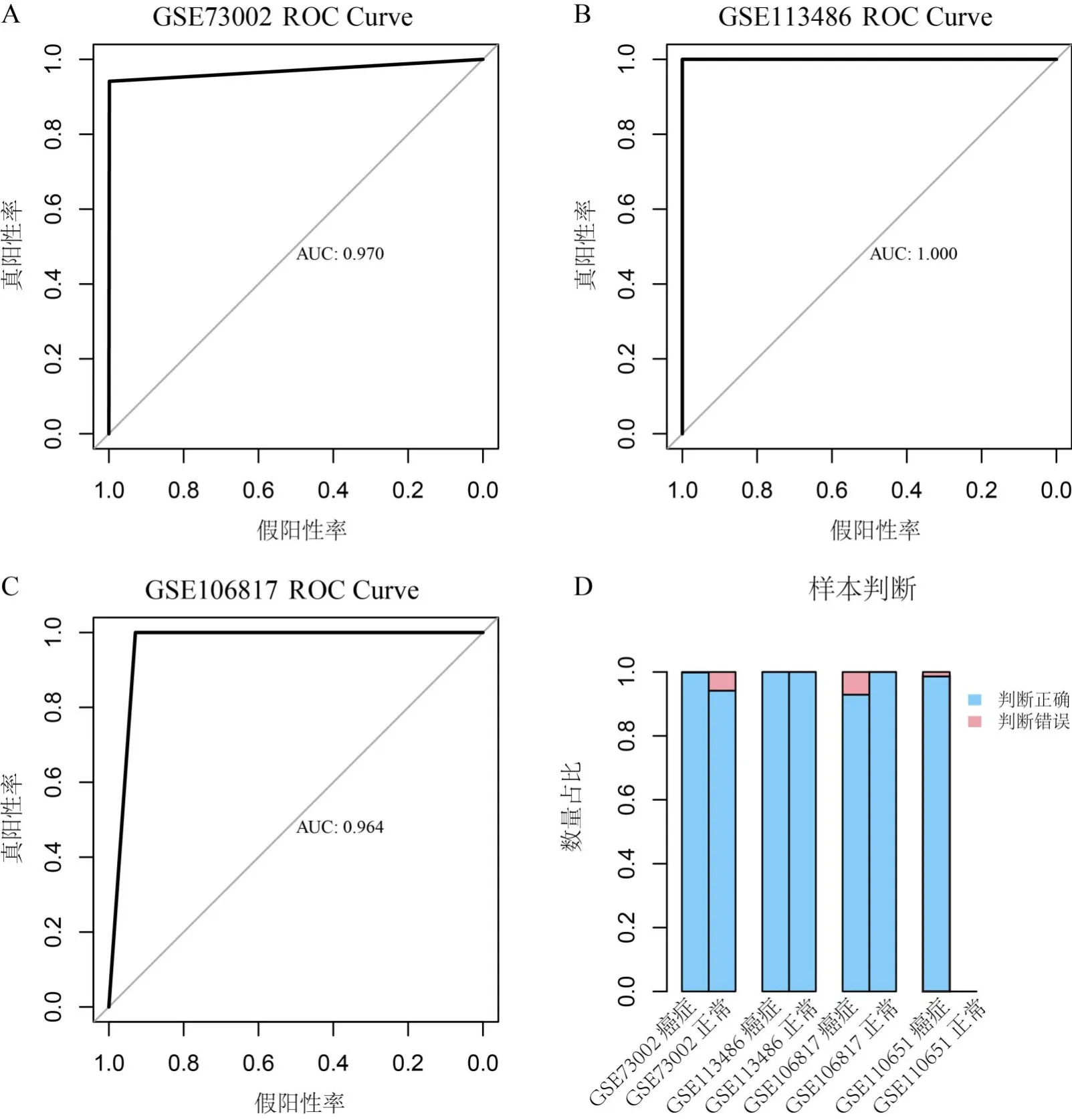
图3 训练集及验证集的ROC曲线及样本判别情况
2.3 共表达网络分析为了进一步分析预警标志,利用mirDIP 数据库寻找13 个miRNA 的靶基因。在筛选条件miRNA 得分值前1%的靶基因下有4 个miRNA 未找到靶基因,因此选取了这4 个miRNA 的前5%的靶基因和其余9 个miRNA 得分前1%的靶基因。最终获得324 个靶基因,绘制miRNA-靶基因互作网络图(图4A)。利用DAVID 对这些靶基因进行GO(Gene Ontology)和KEGG(Kyoto Encyclopedia of Genes and Genomes)富集分析。靶基因主要参与调控的通路有PI3K-Akt 信号通路[22],细胞增殖的正向调控、转录、RNA 聚合酶Ⅱ启动子的转录调控等通路(图4B)。这些功能可能参与乳腺癌的发生发展。利用String 蛋白质数据库(https://www.stringdb.org/)筛选蛋白质的互作关系,构建蛋白质互作网络。去除孤立点并过滤低可信度的相互作用后,通过网络拓扑学分析和K-均值聚类算法,蛋白质聚合为四个功能模块(图4C)。对四个功能模块的基因进行GO 和KEGG 功能富集,按照图4C 所示排布顺序,结果如图5所示。蓝色模块主要参与RNA 聚合酶Ⅱ启动子的转录及转录的正向调控、转录因子活性、序列特性DNA 结合、DNA 模板等通路(图5Aa);红色模块主要参与蛋白质结合、蛋白质激酶活性调控、线粒体膜通透性调节等通路(图5Ab);黄色模块主要参与mRNA 剪接、剪接位点选择和加工、RNA 结合、DNA 结合和mRNA 结合等通路(图5Ac);绿色模块参与神经嵴细胞迁移、交感神经元投射引导与延伸、神经元投射引导等通路(图5Ad)。KEGG 富集分析未得出显著有效的通路,如图5B 所示。分析结果进一步印证了该预警标志潜在的预测能力。
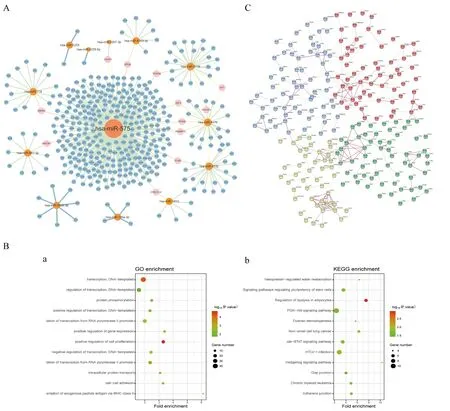
图4 miRNA-靶基因网络图、富集分析图、蛋白质互作网络图

图5 功能模块富集分析图
3 讨 论
本文采用样本内miRNA 相对表达秩序关系的方法在血清中筛选与乳腺癌相关的分子改变特征。在正常样本和乳腺癌样本中筛选高度稳定的逆转miRNA 对作为候选标志对。选择前11 对miRNAmiRNA 作为预警标志,对训练集和验证集样本进行验证。样本内miRNA 相对表达秩序关系构建的预警标志对实验批次效应和样本内数据标准化方法不敏感并且对于微量RNA 的扩增偏移和存在不同程度RNA 降解的样本中也可保持稳健性。结果显示,基于样本内相对表达秩序关系构建的预警标志对乳腺癌样本和正常对照样本的预测准确率为93%~100%。表明该标志能够准确判别乳腺癌样本和正常样本,可作为乳腺癌早期诊断的预警标志。
有研究表明,乳腺癌早期患者治愈率比较高,达到了80%~90%[4],但早期患者病症并不明显,容易被忽视,导致患者确诊时就已经处于中晚期。因此需要一种敏感的、非侵入性的分子标志物来提高乳腺癌的早期检测准确率。许多研究报道,基于基因差异表达水平寻找可以对乳腺癌进行早期诊断的生物标志,或分析不同亚型乳腺癌患者总体生存率,选出与生存显著相关的基因作为生物标志[15-17]等。一些研究也将目光转向血清miRNA,并检测血清miRNA 作为生物标志物的潜力[18-19,23-24],大部分研究只是单纯的研究单个miRNA 的表达对疾病的影响,但miRNA 表达水平容易受多种因素影响。而本研究利用样本内miRNA 相对表达秩序关系的方法构建的预警标志对实验批次效应和样本内数据标准化方法不敏感[25],并且对于微量RNA 的扩增偏移[26]和存在不同程度RNA 降解[27]的样本中也可保持稳健性,因此构建的预警标志更稳健有效。
本文从血清样本中寻找到一种敏感的、非侵入性的生物标志来提高乳腺癌的早期检测准确率,该生物标志能够很好地将乳腺癌样本和正常样本进行判别。然而筛选构建的miRNA-miRNA 标志只在4 套数据集中进行验证,未来还需要在更多的临床样本中进行验证,以便进一步研究。
