利福昔明在人工胃/肠液和大鼠肠道菌群及组织样品中的稳定性初步分析
2022-05-17刘希望白莉霞李世宏李剑勇杨亚军
陶 琦 刘希望 秦 哲 白莉霞 李世宏 李剑勇 杨亚军
(中国农业科学院 兰州畜牧与兽药研究所/农业农村部兽用药物创制重点实验室/ 甘肃省新兽药工程重点实验室,兰州 730050)
利福昔明是利福霉素衍生物(图1),其抗菌谱广,抗菌活性强,局部给药不易吸收,是高效低毒的抗生素。该药物对革兰氏阳性菌、革兰氏阴性菌、需氧菌和厌氧菌等均有良好的抗菌活性;利福昔明(Rifaximin)是由意大利Alfa Wassermann S.P.A公司研制生产,1987年作为抗感染性腹泻药物在意大利上市,之后在国外被广泛研究与应用;2004年通过SFDA批准,在我国广泛应用于医学临床;近年来亦被用于兽医临床。利福昔明在医学临床中常被用于胃肠疾病的治疗,如炎症性肠病(Inflammatory bowel disease, IBD)、肠易激综合征(Irritable bowel syndrome, IBS)等。在兽医临床中,利福昔明以灌注剂的形式用于动物疾病的治疗。子宫和乳房灌注给药后,可在子宫和乳房内保持较高的药物浓度,以杀灭其中的敏感病原菌,用于相关疾病的治疗,如奶牛子宫内膜炎和乳房炎等。

图1 利福昔明结构式Fig.1 Structure of rifaximin
口服给药是最为便捷的给药途径之一,而目前尚无口服的利福昔明制剂被批准用于兽医临床。口服药物进入体内会受到胃肠不同酸碱环境、消化酶、肠道菌群,以及肠壁、肝脏代谢酶的作用,从而影响其药效的发挥。国内兽医临床上,利福昔明以子宫和乳房灌注给药为主。本研究采用高效液相色谱法(High performance liquid chromatography, HPLC),考察了利福昔明在人工胃/肠液中的稳定性以及大鼠肠道菌群、肠道及肝匀浆等对其的降解作用,为兽用相关口服制剂的设计与开发提供数据支持。
1 材料与方法
1.1 药品与试剂
利福昔明对照品(中国食品药品检定研究院,纯度98.8%,批次:130496-201403),乌拉坦(NJKOCED,批号:C0028),胰蛋白酶(猪源,酶活 250 NFU/mg,索莱宝生物科技有限公司,批号:T8150),胃蛋白酶(猪源,酶活3 000-3 500 NFU/g,索莱宝生物科技有限公司,批号:P8390),磷酸盐缓冲液(索莱宝生物科技有限公司,批号:D1040-500),厌氧培养基(CM1513,北京陆桥技术股份有限公司),乙腈(色谱纯,Fisher),盐酸、磷酸二氢钾、氢氧化钠等均为国产分析纯试剂。
1.2 主要仪器
超高效液相色谱仪-二极管阵列检测器(安捷伦1290),匀浆机(IKA-T25),离心机(Multifuge X3R),厌氧培养箱(Thermo)。
1.3 试验动物
清洁级SD大鼠,6只,雄性,8~10周,体重210~230 g,购于中国农业科学院兰州兽医研究所,动物生产许可证号:NKMYD201907018。在试验开始前,动物经过2周的隔离和适应。所有的试验方案和程序,均获得中国农业科学院兰州畜牧与兽药研究所动物伦理委员会的批准。
1.4 相关溶液配制
1
.4
.1
标准储备液与标准溶液称取利福昔明对照品适量,以乙腈溶解,配制成质量浓度为1 mg/mL的标准储备液,于4 ℃存放备用。使用前以乙腈稀释,得到实验所需质量浓度的标准溶液。
1
.4
.2
人工胃液的制备按文献方法[17],取稀盐酸16.4 mL,然后加水800 mL和胃蛋白酶10 g,充分溶解后,用3.65 g/L盐酸溶液调节pH至1.3后,加水稀释并定容至1 000 mL,即得人工胃液。空白人工胃液的配制:不含胃蛋白酶,其余与人工胃液配制方法相同。
1
.4
.3
人工肠液的制备按文献方法[17],取磷酸二氢钾6.8 g,加水500 mL使其溶解,用4 g/L氢氧化钠溶液调节pH至6.8;称取胰蛋白酶10 g,加适量水使其充分溶解;将上述两种溶液混合后,再以水稀释并定容至1 000 mL,即得人工肠液。空白人工肠液的配制:除不含胰蛋白酶,其余与人工肠液配制相同。
1
.4
.4
SD大鼠肠道菌群的制备,按参考文献[18-19],SD大鼠禁食12 h后,刺激腹部以采集新鲜粪便。将粪便与生理盐水按比例(1 g∶5 mL)涡旋混合制成悬浊液,4 500 r/min离心10 min(4 ℃),再将上清液与厌氧培养液按比例(V
∶V
=1∶9)混合,置厌氧培养箱中37 ℃过夜,制得肠道菌群培养液待用。1
.4
.5
空白组织样品的制备按参考文献[20-22],取SD大鼠(230~250 g) 6只,禁食不禁水过夜,腹腔注射乌拉坦麻醉后,迅速处死,采集肝脏、胃、小肠和大肠,用预冷的磷酸盐缓冲液(4 ℃放置2 h)冲洗表面后剪碎,再将各组织与磷酸盐缓冲液按质量与体积比例(1 g∶5 mL)混合,置于匀浆机中均质后,4 500 r/min离心10 min(4 ℃),取上清液备用。离心前的操作均在冰浴条件下进行。
1
.4
.6
厌氧培养基的制备依培养基使用说明,称取厌氧培养基9.5 g,加水800 mL摇匀,然后加水稀释并定容至1 000 mL,混匀后分装于试管中,于高压湿热条件下灭菌,制成厌氧液体培养基(GAM),4 ℃保存备用。
1.5 利福昔明的HPLC检测方法
1
.5
.1
色谱条件参考文献[23],略有调整。Phenomenex luna C18色谱柱(150 mm×4.6 mm, 5 μm);流动相(A)为3.16 g/L甲酸铵,(B)为V
∶V
=1∶1,等度洗脱(V
∶V
=37∶63);柱温40 ℃;检测波长276 nm;流速1.4 mL/min;进样量20 μL;运行时间10 min。1
.5
.2
特异性分析分别取空白人工胃液(1 mL)、空白人工肠液(1 mL)、人工胃液(1 mL)、人工肠液(1 mL)、人工胃液(1 mL)+利福昔明储备溶液(10 μL)、空白人工肠液(1 mL)+利福昔明储备液(10 μL)、空白人工胃液(1 mL)+利福昔明储备液(10 μL)、空白肠道菌液(1 mL)、空白肝匀浆液(1 mL)、空白胃匀浆液(1 mL)、空白大肠及内容物的匀浆液(1 mL)、空白小肠及内容物的匀浆液(1 mL)。按照“1.7”项下的样品前处理方法操作后,按“1.5.1”项下的色谱条件进样分析,对比色谱图。观察在利福昔明标准溶液的出峰位置处,是否有其他物质干扰。
1
.5
.3
重现性配制质量浓度为10、40和100 μg/mL利福昔明标准溶液各6份,再按“1.5.1”项下进样1次,记录峰面积。
1.6 重复性
配制质量浓度为10、40和100 μg/mL利福昔明标准溶液各1份,于不同时间点(0、4、8、12和24 h)按照 “1.5.1”项下条件进样分析,记录峰面积。
1.7 利福昔明的稳定性分析
1
.7
.1
利福昔明在人工胃/肠液中的稳定性测定将上述制备好的空白人工胃液、空白人工肠液、人工胃液、人工肠液迅速分装到EP管中(每管1 mL,每个时间点平行3份),加入利福昔明储备液(1 mg/mL)10 μL,混匀后于37 ℃水浴中孵育不同时间后分别取样300 μL,加入冰乙腈(-20 ℃放置2 h)700 μL,涡旋混匀以终止反应,于4 ℃条件下12 000 r/min 离心10 min,取上清液过0.22 μm滤膜后,按“1.5.1”项下色谱条件进样分析,记录色谱峰面积。
1
.7
.2
利福昔明在SD大鼠肠道菌群中的稳定性测定移取过夜培养液1 mL于EP管中,分别加入利福昔明标准储备液10 μL,涡旋混匀后置入厌氧培养箱,培养不同时间后分别取样0.5 mL于EP管中(每个时间点平行3份),加入冰乙腈1 mL,涡旋混匀以终止反应,11 000 r/min离心10 min,取上清液过0.22 μm滤膜后,按“1.5.1”项下色谱条件进样分析,记录色谱峰面积。以培养相同时长不含利福昔明的培养液为空白对照,同法操作。
1
.7
.3
利福昔明在空白组织样品中的稳定性测定将上述制备好的上清液迅速分装到EP管中(每管1 mL,每个时间点平行3份),加入利福昔明标准储备液10 μL,混匀后置37 ℃水浴中孵育不同时间后,移取300 μL,加入700 μL冰乙腈,充分涡旋以终止反应,12 000 r/min离心10 min,取上清液过0.22 μm滤膜后,按“1.5.1”项下色谱条件进样分析,记录色谱峰面积。
1.8 数据分析
采用GraphPad Prism 8软件进行数据的统计分析,数据以Mean±SD表示。P
<0.05表示差异显著,P
<0.01表示差异极显著。2 结果与分析
2.1 高效液相色谱法(HPLC)测定利福昔明的特异性分析
如图2所示,在前述色谱条件下,利福昔明可被检出,保留时间均为5.45 min,分离良好,不受背景干扰。表明在前述的样品处理及色谱条件下,利福昔明的检测不受基质干扰。

(a)空白人工胃液;(b)空白人工胃液+10 μg/mL利福昔明;(c)人工胃液;(d)人工胃液+利福昔明;(e)空白人工肠液;(f)空白人工肠液+10 μg/mL利福昔明;(g)人工肠液;(h)空白大鼠菌液;(i)空白大鼠大肠及内容物;(j)空白大鼠小肠及内容物;(k)空白大鼠胃液;(l)空白大鼠肝液。利福昔明保留时间为5.45 min。
2.2 高效液相色谱法(HPLC)测定利福昔明的重现性分析
如表1和2所示,利福昔明低、中、高3种质量浓度在不同时间点所得到的峰面积和保留时间。峰面积的RSD值为分别为0.27%、0.26%和0.25%;保留时间的RSD值分别为0.67%、0.33%和0.13%。结果表明方法重现性良好。
2.3 高效液相色谱法(HPLC)测定利福昔明的重复性分析
检测相同浓度的平行样品得到的峰面积如表3所示。RSD 值为分别为0.22%、0.16%和0.26%,表明方法的重复性良好。
2.4 利福昔明在人工胃/肠液中的稳定性
以0 h时利福昔明的峰面积为100%,计算各时间点的剩余百分比,结果见图3。如图所示,经过37 ℃孵育6 h后,在空白胃液、人工胃液及空白肠液、人工肠液中的相对剩余百分率分别为(91.57±1.29)%、(84.03±0.49)%、(81.14±1.69)%和(78.12±1.20)%。尽管空白人工胃液4 h和6 h孵育后的剩余百分比分别比3和5 h的稍高,但没有显著性差别且基本处于稳定状态,主要原因可能是测定误差造成的。结果说明,利福昔明在碱性或消化酶存在的条件下更易降解。
表1 HPLC测定利福昔明时仪器响应的稳定性分析
Table 1 Stability analysis of instrument response for determination of rifaximin by HPLC
质量浓度/(μg/mL)Concentration峰面积/(mAU·min) Chromatographic peak area0 h4 h8 h12 h24 h平均值Average标准差SD相对标准差/%RSD10171.1171.5171.1170.7171.9171.30.460.2740610.6608.1606.6607.1607.4608.01.570.261001 452.61 443.21 145.51 444.71 446.81 446.63.620.25
表2 HPLC测定利福昔明时仪器保留时间的稳定性分析
Table 2 Stability analysis of instrument retention time for determination of rifaximin by HPLC
质量浓度/(μg/mL)Concentration保留时间/min Retention time0 h4 h8 h12 h24 h平均值Average标准差SD相对标准差/%RSD105.3555.4495.4465.3845.4025.4070.0360.67405.4505.4595.4985.4505.4535.4620.0180.331005.4315.4285.4255.4295.4465.4320.0070.13
表3 HPLC测定利福昔明的重复性分析
Table 3 Repeatability analysis of rifaximin determination by HPLC
质量浓度/(μg/mL)Concentration峰面积/(mAU·min) Chromatographic peak area123456平均值Average标准差SD相对标准差/%RSD10171.1171.2170.6171.3171.6170.7171.10.380.2240608.7606.7608.1607.9608.2609.6608.10.950.161001 442.61 446.71 444.61 445.91 452.814 50.71 447.23.830.26

图3 利福昔明在人工胃肠液中的孵育后的剩余利百分比Fig.3 The remaining percentage of rifaximin after incubation in artificial gastrointestinal fluid
2.5 利福昔明在SD大鼠肠道菌群中的稳定性
以0 h时利福昔明的峰面积为100%,计算各时间点的剩余百分比,结果见图4。经过37 ℃孵育12 h后,利福昔明在SD大鼠肠道菌群中孵育时发生了部分降解,但剩余百分比仍在90%上,总体表现出稳定的趋势。结果表明,利福昔明不易受SD大鼠肠道菌群的影响。
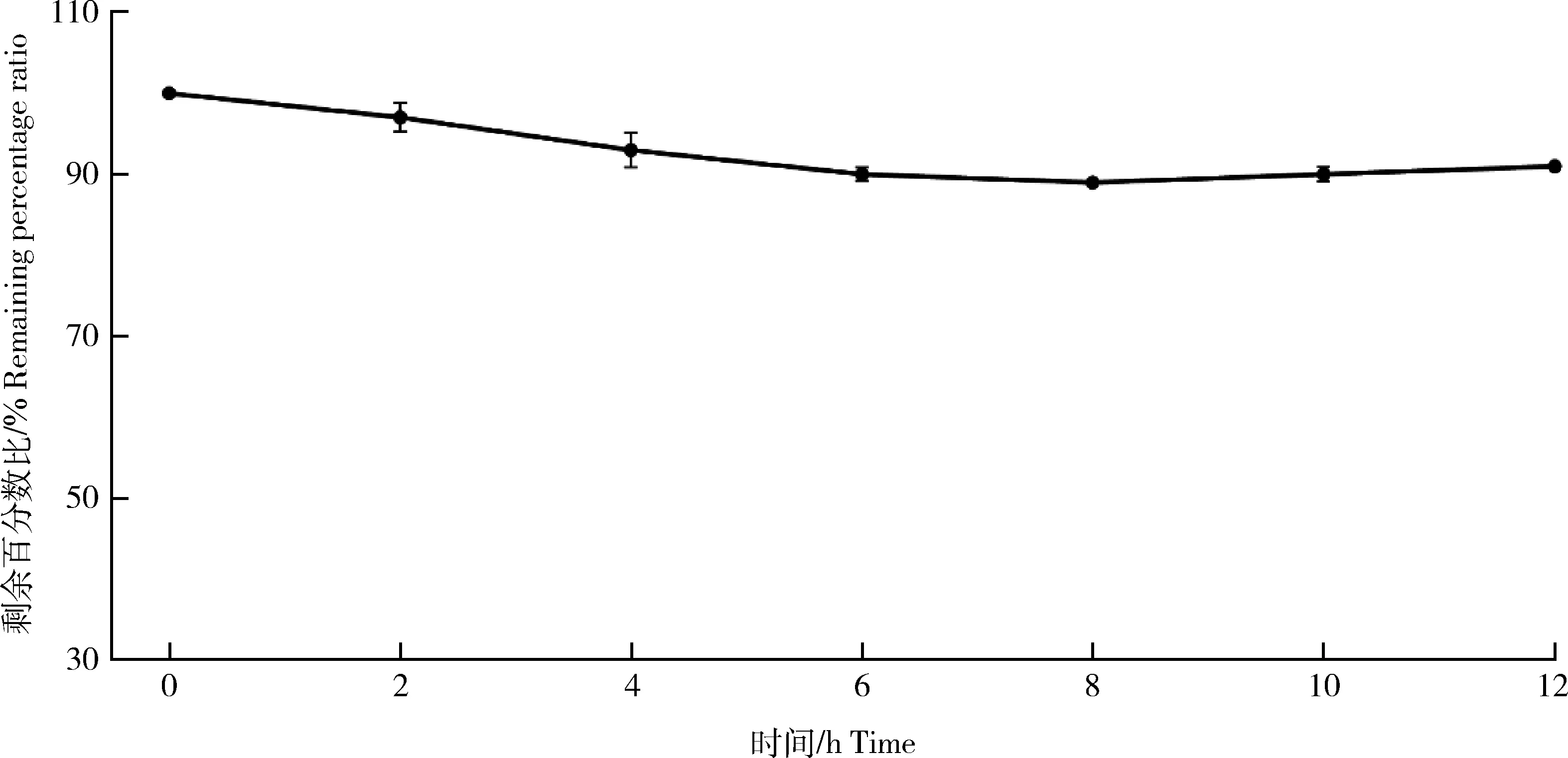
图4 利福昔明在SD大鼠肠道菌群中的孵育后的剩余百分比(n=3)Fig.4 The remaining percentage of rifaximin in the intestinal flora of SD rats after incubation (n=3)
2.6 利福昔明在组织样品中的稳定性分析
以0 h时利福昔明的峰面积为100%,计算各时间点的剩余百分比,结果见图5。如图所示,经37 ℃ 孵育6 h后,利福昔明在胃液、肝液、大肠及其内容物、小肠及其内容物的相对剩余百分率分别为(92.09±0.63)%、(75.95±0.14)%、(83.90±0.91)%和(78.24±0.35)%。结果表明,利福昔明在空白SD大鼠胃匀浆液中基本稳定,但在空白SD大鼠肝匀浆液、大肠及内容物匀浆液、小肠及内容物匀浆液中有一定的降解。
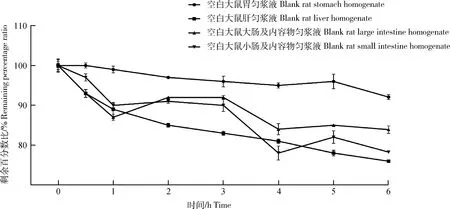
图5 利福昔明在生物样品中孵育后的剩余百分比(n=3)Fig.5 The remaining percentage of rifaximin after incubation in biological samples (n=3)
3 讨 论
口服给药是最为便捷的给药方式,药物在胃肠道的稳定性,直接影响其药效的发挥。因此,对药物在胃肠道环境下的稳定性进行考察,是新药制剂研究开发的重要内容。
利福昔明是半合成的利福霉素类抗菌药物,抗菌活性强,作用范围广,口服后不易吸收,可在肠道局部高浓度环境而发挥抗菌优势。在兽医临床中,利福昔明仅以注入剂的形式,被用作奶牛子宫内膜炎和乳房炎的治疗。体外条件下考察胃肠液、肠道菌群、肠道及肝脏匀浆液对其稳定性的影响,有助于利福昔明兽用口服制剂的研究与开发。
人工胃肠液模型包括人工胃液和人工肠液,肠道菌群可直接代谢药物或作用于宿主系统,间接影响药物代谢,动物组织样品匀浆液更加接近体内环境;此三种体系易于制备,可在一定程度上反映体内情况,以此考察药物的稳定性,有助于了解药物在胃肠道内的稳定性情况,探讨影响药物稳定性的因素,有助于指导药物结构优化和制剂的开发,在药物研究与开发中已经被广泛应用。
因此,本研究将利福昔明分别与人工胃肠液、SD大鼠(雄性)肠道菌群及不同组织样品的匀浆液等共孵育,以考察pH、消化酶、肠道菌群和组织中的酶,对其稳定性的影响。试验结果说明:在体外模拟的胃肠道环境中,利福昔明是较稳定的药物,在酸性的空白人工胃液(不含酶)中总体呈现稳定趋势,而在碱性的人工肠液中有一定的降解,这可能与利福昔明的结构有关。肝脏是药物代谢的主要器官,其中肝脏药物代谢酶发挥主要的作用。利福昔明在SD大鼠肝匀浆液中有一定的降解,说明肝脏药物代谢酶对利福昔明有一定的生物转化作用;利福昔明在空白大鼠大肠及内容物匀浆液、小肠及内容物匀浆液中均有一定的降解,说明肠道对其稳定性有一定的影响。Descombe等对利福昔明做了人体药动学研究,结果显示,志愿者口服给药48 h后,一半以上血浆样本未检测出利福昔明,在给药4 h后,仅有一半的血浆中能检测出极少量的药物(2~5.3 ng/mL)。在给药后48 h,尿液样本中检测出微量的原形药物;旅行者腹泻患者每天口服利福昔明800 mg,连续口服3 d后,粪便中的原型药物浓度非常高,平均为7 961 μg/g。研究结果表明,利福昔明在体内主要以原形发挥作用,并通过粪便以原形排出体外。说明利福昔明在胃肠道和肝脏内的降解较少。本研究结果亦表明,利福昔明在不同条件下仅发生了少量降解,与文献报道基本一致。综上,利福昔明在酸性环境中较稳定,而在碱性条件下,胰蛋白酶、胃蛋白酶和肠壁、肝脏代谢酶等对其稳定性的影响亦较为有限;因此,在开发兽用利福昔明口服制剂时,可减少对药物本身稳定性的考虑。后续将进行体外Caco-2细胞试验,分析结肠上皮细胞对利福昔明的吸收转运情况,为利福昔明开发兽用口服制剂提供数据支持。
