表柔比星固体脂质纳米粒的制备及透黏膜行为*
2022-05-11李伟泽付丽娜王小宁任菲菲
赵 宁,李伟泽,付丽娜,王小宁,马 秀,任菲菲
(西安医学院 药学院,陕西 西安 710021)
表柔比星(Epirubicin hydrochloride,EPI)为蒽醌类抗肿瘤药,具有广谱、高效的特点,主要用于乳腺癌、恶性淋巴瘤、软组织肉瘤和胃癌等的治疗。因表柔比星原药在外部环境下不稳定,市售表柔比星一般以其盐酸盐形式存在。EPI对不同生长周期的细胞均可产生抑制DNA、RNA和蛋白合成的细胞毒作用。目前中国使用的剂型以冻干粉和水针剂为主,但常规制剂存在靶向性差的弊端,不能选择性地区分肿瘤细胞和正常细胞,不良反应较多,存在严重的心脏毒性和骨髓抑制作用。其次,透生物膜能力差,生物利用度低,这些缺点一定程度限制了其临床应用[1-2]。
固体脂质纳米粒(Solid lipid nanoparticles,SLNs)是一种新型的实体骨架微粒给药系统,粒径约为20~1 000 nm。通常以室温下固态的天然或合成的脂质、类脂为基质,将药物包裹于类脂核中,与传统的脂质体相比,有效解决了药物容易泄漏的缺点,同时也避免了载体的物理不稳定性。因其所用基质多为人体耐受性良好的脂质材料,具有良好生物相容性和生物可降解性,且能有效提高药物的生物利用度,增加药物在体内的吸收和靶向性,延缓药物释放。同时SLNs成本低,利于大规模生产,目前已成为微乳、脂质体、聚合物纳米粒等的替代品[3-5],因此具有良好应用前景。作者研究了以复乳法制备EPI-SLNs的处方及成型工艺,并考察了EPI-SLNs体外释药及透黏膜行为。
1 实验部分
1.1 试剂与仪器
盐酸表柔比星原料药:质量分数≥98%,大连美仑生物技术有限公司;盐酸表柔比星标准品:批号130560-200501,中国食品药品检定研究院;大豆卵磷脂:上海太伟药业有限公司;单硬脂酸甘油酯、硬脂酸:南昌华鑫医药化工有限公司;普朗尼克F-68:天津市科密欧化学试剂有限公司;胆固醇:安徽科宝生物工程有限公司;胆酸钠:东京化成工业株式会社;聚氧乙烯40单硬脂酸酯:江苏省海安石油化工厂;色谱乙腈、色谱甲醇:美国Honeywell;其他试剂均为分析纯,市售;水:双蒸水,自制。
高效液相色谱仪:Agilent1260,美国安捷伦科技有限公司;扫描电镜:ZEISS Gemini 300,德国蔡司公司;透射电镜:JEM-2100F,日本电子株式会社;激光粒度仪:ZEN-3600,英国马尔文公司;离心机:Allegra 64R Centrifuge,美国BECKMAN公司;分析天平:Quintix224-1CNsartorius,赛多斯科学仪器北京有限公司;高速分散匀质机:FJ-200,巩义市予华仪器有限责任公司。
1.2 实验方法
1.2.1 EPI含量测定方法的建立
采用高效液相色谱仪,在色谱柱Eclipse XDB-C18(4.6 mm×150 mm,5 μm),流动相为V(乙腈)∶V(水)=37∶63,流速为1.0 mL/min,柱温为30 ℃,检测波长为233 nm,进样量为20 μL的色谱条件下,对制剂中EPI含量进行检测,建立标准曲线,并进行方法学考察[6]。
1.2.2 EPI-SLNs的包封率测定方法
采用冷冻高速离心法测定包封率,精密吸取制好的EPI-SLNs 2 mL,经高速冷冻离心机,t=10 ℃、n=13 500 r/min离心60 min,取上清液1 mL置于2 mL容量瓶中,甲醇定容,经0.22 μm微孔滤膜过滤,取滤液20 μL,测定游离EPI药量,包封率计算见式(1)。

(1)
1.2.3 EPI-SLNs的制备
(1)制备方法。制备固体脂质纳米粒的方法较多,有复乳法、微乳超声法、高速匀质法等,经前期预实验筛选,以复乳法所得固体脂质纳米粒粒径、电位及包封率较好,因此实验以复乳法制备表柔比星固体脂质纳米粒。具体制备步骤如下。称取11 mg单硬脂酸甘油、3.5 mg大豆卵磷脂置于25 mL烧杯中加入3 mL乙酸乙酯作为油相,称取1 mg EPI、5 mg胆酸钠溶于适量的蒸馏水中作为水相,油相、水相分别置于70 ℃水浴中,使其溶解,后将水相缓慢加入油相,涡旋30 s,超声20 s使其形成W/O型初乳,再加入8 mL质量分数为0.1%泊洛沙姆188和质量分数为0.25%卖泽-52中超声90 s得W/O/W复乳,将复乳分散到质量分数为0.1%泊洛沙姆188的4 mL水溶液中,旋转蒸发除去有机溶剂,即得。
(2)EPI-SLNs的处方优化。药物包封率是衡量SLNs制剂质量好坏的重要指标,也是其能够发挥较普通制剂高效、低毒的关键。以SLNs的包封率为评价指标,考察在制备过程中m(单硬脂酸甘油酯)(2、3.5、5、8、11 g),m(大豆卵磷脂)(1.5、2.5、3.5、4.5、5.5 mg),V(油相)∶V(水相)(2、3、4、5、6)对EPI-SLNs包封率的影响。
(3)EPI-SLNs的工艺优化。乳化过程对EPI-SLNs的成型至关重要,考察了制备过程中初乳的涡旋时间(20、30、40、50、60 s)、初乳超声时间(10、20、30、40、50 s)与复乳超声时间(30、60、90、120、150 s)对EPI-SLNs包封率的影响[7-11]。
1.2.4 EPI-SLNs药剂学性质考察
(1)包封率测定。按照上述筛选的最优处方及最佳工艺制备3批EPI-SLNs,其外观均匀一致,t=4 ℃存放,备用。按1.2.2方法测定其包封率。
(2)粒径及电位测定。将按最优处方和工艺制备的EPI-SLNs适量,置于50 mL烧杯中,按照V(EPI-SLNs)∶V(纯净水)=1∶10稀释,经马尔文激光粒度仪测定其粒径及电位[12-14]。
1.2.5 EPI-SLNs的体外释药行为考察
采用平衡透析法考察EPI-SLNs的体外释放特性。取所制备的EPI-SLNs混悬液10 mL置于经处理的透析袋中,两端扎紧,置于50 mL的生理盐水中,于恒温磁力搅拌器上,设定t=37 ℃、n=450 r/min,分别在t=0、1、2、4、8、12、24 h取样1 mL并及时补充等量的生理盐水,样品经0.22 μm微孔滤膜过滤,经HPLC检测,计算EPI的累积释药量。以药物累积释放率为纵坐标,时间为横坐标作图,同法以EPI水溶液为对照[15]。
1.2.6 EPI-SLNs的体外透黏膜行为考察
采用立式扩散池法考察EPI-SLNs的体外透黏膜行为,具体操作如下。将处理好的猪小肠固定于接收池和供给池之间,黏膜侧面向供给池,供给池中加入所制备的EPI-SLNs,接收池中以7.0 mL的生理盐水作为接收液。分别在给药后t=1、2、4、8、12、24、36 h分别精密吸取接收液1.0 mL,并补加等量的新鲜生理盐水,样品经0.22 μm微孔滤膜过滤,取20 μL注入HPLC测定EPI的含量。以EPI水溶液作为对照组,同法操作。各时间点的药物累积透过率见式(2)。以累积透过率为纵坐标,时间为横坐标作图[16-17]。
(2)
式中:Q为药物累计释放质量占总质量的百分数,%;ρi为第i次所取样品的质量浓度,mg/L;V为释放介质总体积,mL;Vi为取样体积,mL;m为药物总质量,mg。
1.2.7 EPI-SLNs的黏膜滞留量考察
在透黏膜实验36 h后,将肠黏膜取下,晾干后称量并剪碎,置于烧杯中,加入8 mL生理盐水,超声30 min,移至10 mL容量瓶中,定容至刻度,转移至离心管中,t=10 ℃、n=13 500 r/min离心30 min,取上清液,过0.22 μm微孔滤膜后,经HPLC测量黏膜中滞留药物的含量。
2 结果与讨论
2.1 EPI含量测定的方法学考察结果
2.1.1 标准曲线的建立
精密称取EPI标准品1.000 mg,置于50 mL容量瓶中,用甲醇稀释至刻度,即得20 μg/mL的EPI储备液,备用。精密吸取储备液0.039、0.078、0.156、0.312、0.625、1.25、2.5 mL分别置于5 mL容量瓶中,加甲醇稀释至刻度,得到质量浓度为7.8、15.6、31.2、62.4、125、250、500 μg/mL的系列标准品溶液,分别标为1~7号,经0.22 μm微孔滤膜,取20 μL,按照1.2.1色谱条件测定,以峰面积A为纵坐标,以质量浓度ρ为横坐标,进行线性回归,得回归方程y=61.415x+90.228,R2=0.999 2,表明ρ(盐酸表柔比星)=7.8~500 μg/mL线性关系良好。
2.1.2 方法学考察
(1)精密度实验。取EPI 3号标准品溶液20μL,按1.2.1色谱条件测定峰面积,连续进样6次,考察精密度,结果峰面积积分值的RSD为0.2%,精密度良好(n=6)。
(2)稳定性实验。取EPI 4号标准品溶液室温下放置,分别于t=0、3、6、12、24、48 h取20 μL注入HPLC,按照1.2.1色谱条件测定其峰面积,峰面积积分值的RSD为0.02%,表明盐酸表柔比星在t<48 h稳定性良好。
(3)重复性实验。精密吸取制好的EPI-SLNs 2 mL,置于2 mL离心管,t=10 ℃、n=13 500 r/min离心60 min,取上清液1 mL置于2 mL容量瓶中,甲醇定容,经0.22 μm微孔滤膜过滤,取20 μL按照1.2.1色谱条件测定峰面积,连续进样6次,考察重复性,结果峰面积积分值的RSD为1.61%,重复性良好(n=6)。
(4)回收率实验。取9份空白固体脂质纳米粒1 mL,置于2 mL离心管内,分为3组,分别加入62.5、125 L、250 μg/mL EPI标准品溶液1 mL,t=10 ℃、n=13 500 r/min离心30 min,取上清液,过0.22 μm微孔滤膜,进HPLC按照1.2.1色谱条件测定,测得平均回收率为98.8%,峰面积积分值的RSD为2.91%。
2.2 EPI-SLNs的处方及工艺优化结果
2.2.1 EPI-SLNs的处方优化结果
单硬脂酸甘油酯是制备SLNs的主要脂质材料,其用量对SLNs的质量具有重要影响,见图1(n=3)。

m(单硬脂酸甘油酯)/mg
由图1可知,当m(单硬脂酸甘油酯)=5 mg,EPI-SLNs的包封率为62.5%,此后包封率随着m(单硬脂酸甘油酯)增加而降低;推测原因可能为在SLNs的制备过程中,药物与脂质存在一定的容纳比例,因此通过增大脂质材料的用量,提高纳米粒对药物的包封率具有一定限度,且在制备中发现当处方中m(单硬脂酸甘油酯)过大,形成的SLNs混悬液中存在脂质析出的现象。
大豆卵磷脂对于初乳的形成至关重要,影响到SLNs的成型性及包封率,见图2(n=3)。

m(大豆卵磷脂)/mg
由图2可知,随着m(大豆卵磷脂)的增加,EPI-SLNs的包封率呈现先上升后缓慢降低的趋势,m(大豆卵磷脂)=3.5 mg,包封率达到最大为65.9%,推测当m(大豆卵磷脂)较小时,不能够完全乳化硬脂酸甘油酯,故包封率较小,m(大豆卵磷脂)=3.5 mg,体系中磷脂已足够乳化油相,形成稳定的初乳,因此当m(大豆卵磷脂)继续增大,包封率不再明显增大,且当体系中大豆卵磷脂浓度过大,可能形成较多的大小不一的磷脂囊泡,也将影响SLNs的质量及外观形态。
研究表明,当油相组成、载体材料浓度确定后,V(油相)∶V(内水相)也会影响SLNs的包封率,见图3(n=3)。
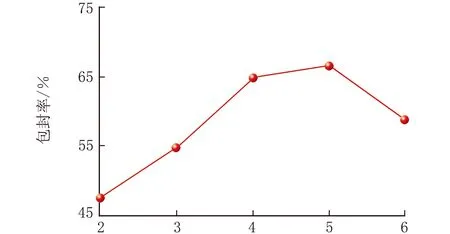
V(油相)∶V(内水相)
由图3可知,EPI-SLNs的包封率随着V(油相)∶V(内水相)的增加呈先上升后下降的现象,当V(油相)∶V(内水相)=5,包封率达到最大为66.7%,在实验中也发现当V(油相)∶V(内水相)较小时,制得的SLNs易胶凝,当V(油相)∶V(内水相)过大时,制得的SLNs混悬液有药物结晶析出。
2.2.2 EPI-SLNs的工艺优化结果
乳化过程对EPI-SLNs的成型至关重要。考察了制备过程中初乳的涡旋时间、初乳超声时间与复乳超声时间对EPI-SLNs包封率的影响,结果见图4~图6(n=3)。

初乳涡旋时间/s
由图4可知,制备初乳时,涡旋30 s,SLNs包封率为69.3%,之后随着涡旋时间的延长,包封率下降,推测原因为涡旋是将体系快速混合,为乳化的准备步骤,其所提供的能量不足以形成均匀稳定的乳剂,因此涡旋时间过长,导致体系中油相、内水相的不完全乳化将不利于后续操作。
由图5可知,初乳超声时间为40 s,SLNs包封率为69.5%,继续延长超声时间,包封率下降,推测原因为超声40 s时初乳已形成,此时体系中乳滴密度较大,继续提供较大能量将导致乳滴的碰撞、合并,影响药物包封率。
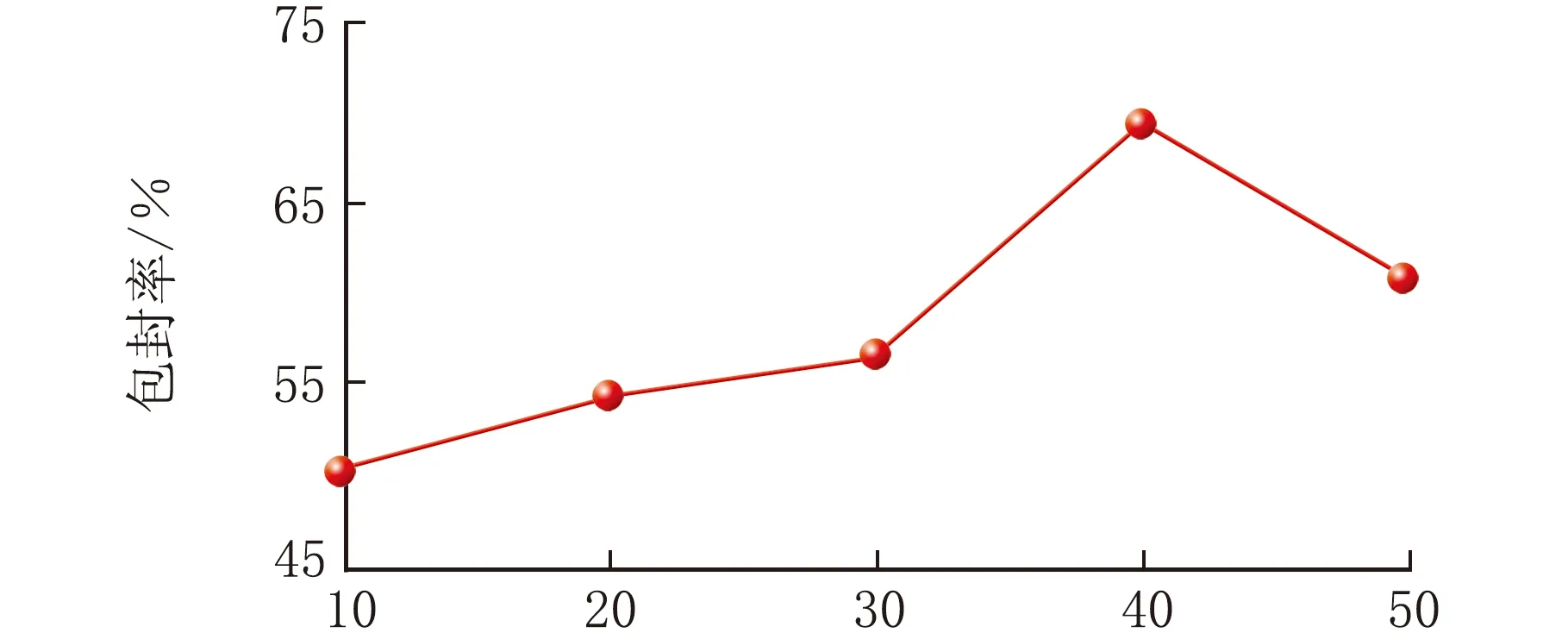
初乳超声时间/s
由图6可知,复乳超声时间为60 s,SLNs包封率为72.3%,继续延长超声时间,包封率降低,推测原因为此时复乳已经形成,若超声时间过长,将易破坏复乳结构,引起内水相的外溢,因此影响包封率。

复乳超声时间/s
综上,得到最佳制备处方及工艺为m(单硬脂酸甘油酯)=5 mg、m(大豆卵磷脂)=3.5 mg,V(油相)∶V(内水相)=5,初乳涡旋时间30 s,初乳超声时间40 s,复乳超声时间60 s。
2.3 EPI-SLNs药剂学性质考察结果
2.3.1 EPI-SLNs的包封率
按照筛选的最优处方及最佳工艺制备3批EPI-SLNs,其外观均匀一致,t=4 ℃存放、备用。测定其包封率,结果见表1。

表1 包封率测定结果
2.3.2 EPI-SLNs的粒径及电位
取最优处方和工艺制备的EPI-SLNs适量,置于50 mL烧杯中,按V(EPI-SLNs)∶V(纯净水)=1∶10稀释,经马尔文激光粒度仪测定,其平均粒径为148.6 nm,粒径PDI为0.259。粒径分布均匀,平均电位为-30.8 mV,一般而言,当粒子表面电位的绝对值大于30 mV时,由于粒子表面存在大量净电荷,纳米粒子之间存在较大的静电斥力而有利于制剂的稳定性,见图7、图8。

d/nm

U/mV
2.4 EPI-SLNs的体外释药行为考察结果
采用平衡透析法考察EPI-SLNs的体外释放特性,见图9(n=3)。
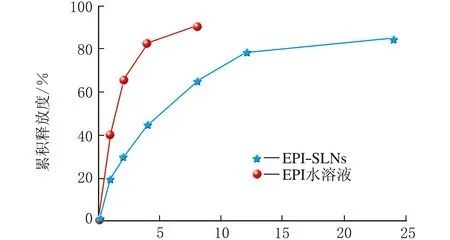
t/h
由图9可知,t=8 h EPI水溶液的释放度已达90%,高于EPI-SLNs,EPI-SLNs呈现明显的缓释作用。EPI-SLNs的体外释药在初期存在突释现象,t=1 h体外累积释放度达到了20.17%,t=4 h为45.02%,推测该阶段释放较快与药物在SLNs中的分布有关,在EPI-SLNs的制备中,部分药物可能以分子或细小粒子形式分布于纳米粒骨架结构的表层或浅表层,而未进入内核,这些药物用药可较快释放,表现为突释。此后EPI释放逐渐缓慢,t=8 h累积释放度达65.02%,该阶段主要是脂质基质逐渐被释放介质溶蚀,药物则通过扩散缓慢从刚性基质结构中释放,故显示明显的缓释。
2.5 EPI-SLNs的体外透黏膜行为考察结果
采用立式扩散池法考察EPI-SLNs的体外透黏膜行为,见图10(n=3)。
由图10可知,EPI-SLNs与其水溶液相比黏膜渗透能力具有明显优势,t=8、12、36 h,EPI-SLNs的黏膜累积透过率分别为48.4%、55.12%、62.08%,为EPI水溶液的2、1.96、1.79倍,表明EPI-SLNs具有更好的黏膜透过性,因此可推测制备成EPI-SLNs后大大提高了其黏膜渗透能力,有利于药物在体内的吸收,在一定程度上可提高EPI的生物利用度。
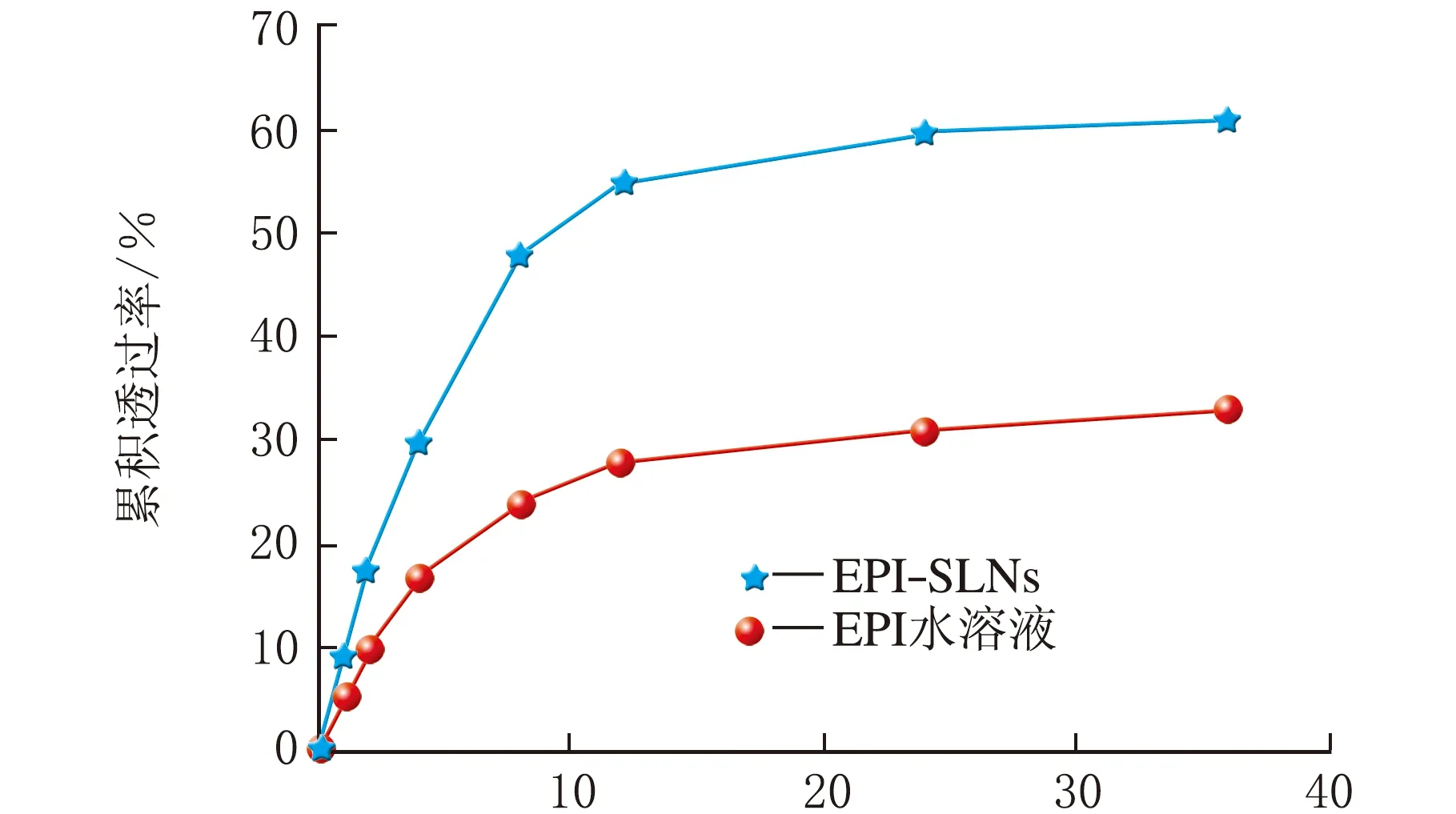
t/h
2.6 EPI-SLNs的黏膜滞留量考察
透黏膜实验进行36 h后,将肠黏膜取下,按1.2.6方法测量黏膜中滞留的药物含量,见图11(n=3)。
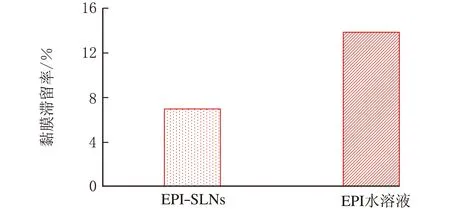
图11 EPI、EPI-SLNs黏膜滞留率
由图11可知,36 h后EPI-SLNs和EPI水溶液的的黏膜滞留量分别为给药量的6.97%和13.84%。结合透黏膜行为可知,EPI-SLNs比EPI水溶液更易透过黏膜,而不易在黏膜组织中滞留,在一定程度上可推测EPI-SLNs体内用药将更利于吸收而毒副作用更小。
3 结 论
制备SLNs常用的脂质材料有单硬脂酸甘油酯、硬脂酸、棕榈酸、胆固醇等,作者在前期预实验中对脂质材料进行了筛选,通过复乳法,以包封率为指标,分别以上述4种材料制备了EPI-SLNs,所得EPI-SLNs包封率分别为59.5%、49.3%、53.8%、55.2%,且以硬脂酸及棕榈酸为脂质材料制备的SLNs混悬液中常出现载体析出的现象,考虑其包封率较低与载体析出及脂质胶凝有关,故实验以单硬脂酸甘油酯为制备SLNs的主要脂质材料。SLNs为纳米级别给药体系,为了得到纳米级别的粒子,制备过程中需要给予足够的能量,将脂质材料有效的分散,通常采用超声或高速搅拌。采用复乳法制备了EPI-SLNs,体外释药行为显示EPI-SLNs具有明显的缓释效果,体外透黏膜实验显示EPI-SLNs相较于其水溶液具有更强的黏膜渗透能力,因此,在一定程度上可预测EPI-SLNs相较于传统制剂,在体内发挥药效更加持久,且更有利于药物的吸收以及生物利用度的提高,因此EPI-SLNs具有广阔的应用前景。
