胶砂比对低钙固碳胶凝材料砂浆碳化硬化性能的影响*
2022-05-09畅祥祥刘松辉房晶瑞刘姚君赵松海管学茂
畅祥祥,刘松辉,张 程,房晶瑞,刘姚君,赵松海,管学茂
(1. 河南理工大学 材料科学与工程学院,河南 焦作 454003;2. 中国建筑材料科学研究总院有限公司,绿色建筑材料国家重点实验室,北京 100024;3. 河南强耐新材股份有限公司,河南 焦作 454950)
0 引 言
混凝土的用量仅次于水,是用量最大的建筑材料[1-2]。普通硅酸盐水泥是混凝土的主要组成材料,混凝土大量的使用,促使普通硅酸盐水泥的产量高居不下,全球年产量超过40亿吨[3]。但是,水泥生产是一个高能耗的过程,水泥中钙含量高是水泥生产过程中能耗高、废气排放大的主要原因。因此,开发低钙胶凝材料是一个趋势。
近几年,有关碳化型低钙胶凝材料的研究越来越多。Wang等[4]研究了γ-C2S碳化后抗压强度、碳化程度和微观结构的变化,发现试块的抗压强度和致密度与碳化程度的变化规律是一致的,γ-C2S的碳化产物方解石、硅胶和未碳化的γ-C2S呈层状分布。Chang等[5]研究了加速碳化过程中β-C2S和γ-C2S的强度发展以及微观结构变化,发现碳化后β-C2S的抗压强度高于γ-C2S,两者的碳化产物均为方解石和高聚合硅胶。Shi等[6]开发了一种C3S2-γ-C2S-C2AS体系的低钙胶凝材料,通过加速碳化6 h后抗压强度能够达到46.5 MPa;抗压强度源于碳化产物中的文石、方解石以及高度聚合的硅胶;碳化后的试样孔隙率减小、微观结构变得更加紧密。Hou等[7]设计了一种以C3S2和γ-C2S为主要矿物组成的新型自粉化低碳熟料,制备成砂浆试块,碳化7 d后抗压、抗折强度分别为54.3和13.5 MPa。综上所述,不同低钙胶凝材料净浆的碳化硬化性能、微观结构和物相组成已经取得了丰硕的研究成果,但是,低钙胶凝材料砂浆制品的碳化硬化性能、界面微观结构以及界面微观力学性能尚没有充分的研究,严重制约了低钙胶凝材料的推广应用。
本研究用工业原料石灰石和砂岩在1 275 ℃烧制一种以C2S、C3S2为主,C2AS为辅的低钙固碳胶凝材料。研究了不同胶砂比下,砂浆试块碳化硬化后的力学强度、碳化程度、吸水率等宏观性能,并通过微观测试来分析砂浆界面的微观形貌、微观力学性能、物相组成以及分布,揭示胶砂比对低钙固碳胶凝材料砂浆宏观性能以及界面性能的影响。
1 实 验
1.1 低钙固碳胶凝材料的制备
制备低钙固碳胶凝材料所用的工业原料石灰石和砂岩,由焦作坚固水泥有限公司提供,其化学组成如表1所示。根据前期的探索,按照石灰石78.2%和砂岩21.8%比例进行混合。将混合好的工业原料粉磨至粒径低于80 μm。粉磨好的工业原料与水以10∶1拌合均匀后,把湿混合料移入模具中。将其在6 MPa压力下压制成型,得到直径30 mm、高度为10 mm的生料片[7]。将生料片移入105 ℃烘箱中烘8 h以上,移入高温炉进行煅烧。从室温以10 ℃/min升温至900 ℃保温0.5 h,接着升温至1 275 ℃保温2 h。将煅烧完成后的熟料放在电风扇下快速冷却至室温。采用乙二醇快速测定游离氧化钙分析方法对烧成的熟料进行游离氧化钙的测定,多次测定取平均值为1.1%,小于1.5%,说明易烧性良好。将烧成的熟料放入球磨机中,球磨50 min后熟料的粒径均小于80 μm。

表1 工业原料的化学成分组成(%质量分数)Table 1 The chemical composition analysis of industrial raw materials (wt%)
1.2 低钙固碳胶凝材料的化学和物理性质
对低钙固碳胶凝材料的XRD图谱进行了全谱拟合(WPF)[8-10]。图1是低钙固碳胶凝材料的Rietveld图,将图中得到的定量分析结果列入表3中。通过对比表2中的设计矿物组成和表3中定量分析得到的实际矿物组成,发现Rietveld物相定量分析各矿物成分含量误差在10%以内,说明低钙固碳胶凝材料矿物组成的实验结果与设计理论是一致的[11]。图2为低钙固碳胶凝材料粉末微观形貌图,从图中可以看出熟料的粒径大约为2~5 μm,形状不规则且表面粗糙。图3是低钙固碳胶凝材料的粒径分布图,由激光粒度分析仪测得低钙固碳胶凝材料的D10为1.90 μm、D50为9.89 μm、D95为74.2 μm,比表面积为1 382 m2/kg。用李氏瓶法测得低钙固碳胶凝材料密度为2.93 g/cm3。

表2 低钙固碳胶凝材料设计矿物组成(%质量分数)Table 2 Design mineral composition of LC-CSB (wt%)

表3 低钙固碳胶凝材料实际矿物组成(%质量分数)Table 3 Actual mineral composition of LC-CSB (wt%)
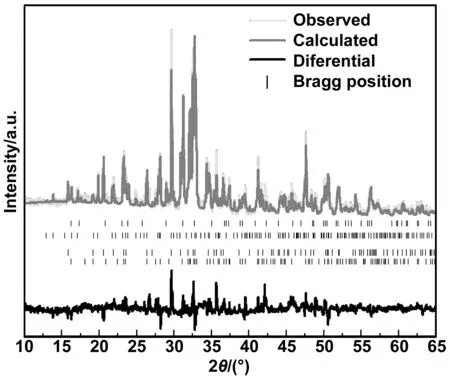
图1 低钙固碳胶凝材料的XRD图谱及Rietveld分析Fig 1 XRD pattern and Rietveld analysis of LC-CSB

图2 低钙固碳胶凝材料粉末微观形貌Fig 2 Micromorphology of LC-CSB powder

图3 低钙固碳胶凝材料的粒径分布Fig 3 Particle size distribution of LC-CSB powder
1.3 样品的制备以及碳化养护
将制备的低钙固碳胶凝材料与标准砂以胶砂比1∶0、1∶1、1∶2、1∶3拌合,拌合的水灰比为0.15。将混合好的湿混合料倒入模具中,在4 MPa的压力下,压制成尺寸为40 mm×40 mm×40 mm的砂浆试块。将砂浆试块转入碳化箱中,向碳化箱中通入纯度99.99%的CO2,直到碳化箱里的压力达到0.3 MPa。在室温条件下,碳化24 h。
1.4 测试方法
1.4.1 碳化程度
碳化程度:对碳化之后的试块进行碳化程度的测试,碳化程度计算公式如式(1)、式(2):
(1)
(2)
其中ω为矿物实际上吸收CO2质量分数,m0为称取矿物的质量,m1为整个装置碳化前充满CO2的质量,mt为整个装置碳化t时刻的质量;ωmax为矿物理论完全碳化吸收的CO2的质量分数。
1.4.2 抗压强度和吸水率
抗压强度:用YAW-300/20型微机控制压力试验机进行试样抗压强度的测试,每次测试3个样品,抗压强度取3次测试的平均值。
吸水率:参考GB/T35160.1—2017合成石材试验方法第1部分:密度和吸水率的测定。
1.4.3 微观性能测试
X射线衍射分析:把测试的试样放在玛瑙研钵中研磨,磨至其粒径小于40 μm,利用日本理学Smart-Lab型X射线衍射仪进行矿物组成分析。靶材为Cu靶,测试过程中的扫描范围10°~70°,扫描速率10°/min。
热重分析:用北京恒久科学仪器厂BJ-HCT-3综合热分析仪进行热分析。测试样品质量约为15 mg,需将样品磨细至40 μm。测试温度范围为25~1 000 ℃,升温速率为10 ℃/min。
红外光谱分析:用德国Bruker公司生产的V70全自动切换傅立叶变换红外光谱仪进行FT-IR测试,采用KBr压片的方法,红外光谱仪的测试范围为400~4 000 cm-1,分辨率为4 cm-1。
纳米压痕:用Triboindenter纳米压痕仪(Nano-indenter)对样品进行测试。在硅质河砂与低钙固碳胶凝材料浆体之间的界面处打点。在8×25的网格中以5 μm为间隔,每个样品打200个点。打点过程中最大荷载为2 000 μN,单个循环加载制度[12]为:加载时间为5 s,保持2 s的恒载时间,卸载时间为5 s。
扫描电镜:采用德国Carl Zeiss NTSGmb H公司Merlin Compact型扫描电镜测试,测试前样品需进行喷金处理。
孔隙率:对充分干燥的样品进行压汞测试,采用的设备为Autopore Ⅳ 9500,最大和最小压力分别为400 MPa和1.4 kPa。通过孔隙率、孔隙分布、孔隙体积和特征孔径等来表征不同胶砂比试块的孔隙结构变化。
2 结果与讨论
2.1 抗压强度和吸水率
不同胶砂比砂浆的抗压强度和吸水率如图4所示,随胶砂比的减小,抗压强度先增大后减小。胶砂比1∶0是不掺砂子的净浆组,抗压强度为28.9 MPa。当胶砂比1∶1时砂浆试块的抗压强度最高为46.9 MPa,相比净浆强度提升62.4%。胶砂比1∶2时砂浆试块的抗压强度降至17.3 MPa,胶砂比为1∶3抗压强度降到7.4 MPa。砂浆的强度呈现先增长后减小的现象。其原因可能为当加入砂子较少时,砂子取代了一少部分胶凝材料,故对试块整体的胶凝性影响不大。但砂子会起到支撑结构的作用,有利于CO2的渗入,促进了碳化反应的进行,从而使砂浆试块的强度增大。当进一步提高砂子的用量时,CO2的渗入速率变化不大,而大量取代胶凝材料的砂子影响到了试块整体的胶凝性,致使抗压强度急剧下降。

图4 不同胶砂比砂浆的抗压强度和吸水率Fig 4 Compressive strength and water absorption of mortar with different cement-sand ratios
砂浆试块的吸水率随胶砂比的减小先减小后逐渐增大。胶砂比1∶1较1∶0吸水率由9.7%降至6.9%。掺入部分砂子,由于砂子的稀释作用,试块的碳化程度变大,内部结构更加致密,吸水率减小。当进一步增加砂子的用量,碳化程度逐渐增大,但低钙胶凝材料的量不足以胶结砂子,致使内部孔隙率变大,吸水率增大。同时内部孔隙变多也是抗压强度急剧下降的主要原因。
2.2 碳化程度
不同胶砂比砂浆试块的碳化程度如图5所示,随胶砂比的减小砂浆的碳化程度先增大然后趋于稳定,胶砂比为1∶0的净浆组的碳化程度为27.51%,胶砂比为1∶1时碳化程度提升到43.0%,继续减小胶砂比碳化程度趋于稳定。胶砂比为1∶0的净浆组结构比较致密,CO2渗入困难,内部碳化不完全,其碳化程度较低。当掺入砂子时,会形成一定支撑结构,有利于碳化反应的进行,故碳化程度高于净浆组。骨料的稀释作用有利于提高CO2的渗透,当胶砂比减小到1∶1时,进一步减小胶砂比对CO2渗入即碳化反应程度影响不大。

图5 不同胶砂比砂浆试块的碳化程度Fig 5 Carbonation degree of mortar with different cement-sand ratios
2.3 碳化产物的组成
2.3.1 碳化产物矿物组成(XRD)
低钙固碳胶凝材料碳化前后矿物组成如图6所示。未碳化矿物组成为β-C2S、γ-C2S、C3S2和C2AS,而在碳化24 h后的XRD图谱中,可以看到方解石(2θ=29.4°、39.44°、43.24°和47.54°)的存在[13],表明低钙固碳胶凝材料碳化后,主要的碳化产物为方解石。且碳化后,β-C2S、γ-C2S、C3S2的衍射峰逐渐减弱,表明β-C2S、γ-C2S、C3S2与CO2发生反应,生成的碳化产物为方解石。而C2AS的衍射峰随碳化时间的增长没有任何变化,表明C2AS没有碳化活性[6]。

图6 低钙固碳胶凝材料碳化前后矿物组成Fig 6 Mineral composition of LC-CSB before and after carbonation
2.3.2 热重分析(TG)
低钙固碳胶凝材料碳化后TG-DTA曲线如图7所示。从图中可以看出,所有样品在100~300 ℃都有质量损失,这部分失重主要是产物中物理、化学结合水蒸发所致[14]。根据TG曲线,可以计算出碳化后的低钙固碳胶凝材料的CO2吸收率为13.82%。更大的质量损失集中在500 ~900 ℃之间,该温度段失重是因为CaCO3分解释放CO2所导致的[15-16],通过失重质量分数可以算出低钙固碳胶凝材料碳化后CaCO3的含量为31.41%。此外,DTA曲线表明500 ~680 ℃之间的失重是结晶度差的CaCO3分解释放CO2引起的,680 ~900 ℃之间的失重则是由结晶度好的CaCO3分解释放CO2引起的[17]。TG-DTA曲线表明主要的失重为CaCO3分解,这与XRD测试碳化后的主要物相为方解石的结论是一致的。

图7 低钙固碳胶凝材料碳化后TG和DTA曲线Fig 7 TG/DTA curves of LC-CSB after carbonation
2.3.3 非晶态产物分析(FT-IR)
低钙固碳胶凝材料碳化前后FT-IR图谱如图8所示。碳化前,在846 cm-1处出现一个强的吸收谱带,与低钙固碳胶凝材料中的C3S2、β-C2S和γ-C2S相对应[18]。碳化后出现了新的峰带(1 410、1 180、1 060、872、712 cm-1)。其中1 410 cm-1处是方解石中C—O键的不对称拉伸(v3),872 cm-1处是C-O键的平面外弯振动(v2)[19],712 cm-1处是方解石的一个弱吸收峰[20]。硅胶中Si-O键峰带范围为900~1 200 cm-1,1 180和1 060 cm-1处是硅胶中Si-O键的主要振动峰带[18]。综合以上测试分析,低钙固碳胶凝材料碳化产物为方解石型CaCO3和高聚合度的硅胶[21-22]。

图8 低钙固碳胶凝材料碳化前后FT-IR图谱Fig 8 FT-IR spectra of LC-CSB before and after carbonation
2.4 孔结构
不同胶砂比砂浆试块碳化后孔径分布如图9所示,随胶砂比减小,累计孔隙率呈现先减小后增大的趋势。胶砂比为1∶1时孔隙率最低,这与吸水率的测试结果是一致的。从孔径分布曲线可以看出砂子掺入后孔径介于0.01 ~1 μm的孔显著减少,大于1 μm孔径的孔增多。这表明了掺入砂子后可以促进碳化反应的进行,碳化产物填充试块内部的小孔,而大孔增多的原因是掺入砂子后,初始试块的密实度变差。对比不同胶砂比孔径分布曲线可以看出孔径介于0.01~1 μm的孔的分布趋势基本是相同,而大于1 μm孔径的孔随砂子掺量的增多逐渐增多。这表明掺入砂子后不同胶砂比试块的碳化程度是一样的,而大孔增多是因为砂子的掺量逐渐增多。砂子掺量增多导致初始试块的密实度逐渐变差,初始孔隙率逐渐的增大,而碳化产物不足以完全填充其全部孔隙,故致使力学性能逐渐变差。

图9 不同胶砂比碳化后砂浆试块孔径分布Fig 9 Pore size distribution of mortar blocks after carbonation with different cement-sand ratios
2.5 界面的微观力学性能
不同胶砂比的低钙固碳胶凝材料砂浆试块碳化后,其胶凝材料基体与砂子界面处的弹性模量云图和平均弹性模量如图10、11、12所示。从图10(a)中可以清晰的看到砂子和胶凝材料基体的界面。界面处和胶凝材料基体均出现中心弹性模量高,往四周延伸弹性模量逐渐降低的等高图样。而未碳化矿物的弹性模量高于碳化产物的弹性模量[3],说明碳化产物的分布状态为碳化产物包裹未碳化矿物[7]。
对比图10(a)、11(a)、12(a)不同胶砂比砂浆试块碳化后界面处的弹性模量云图可以看出,随着胶砂比的减小,胶凝材料基体和界面处未填充色块所代表的弹性模量区域逐渐的增多。该区域所代表的弹性模量与低钙硅酸盐水泥碳化后孔隙的弹性模量[3]基本相同,说明孔隙率的增大导致其弹性模量降低,这与硅酸盐水泥体系的结论是一致的[23]。对比图10(b)、11(b)、12(b)不同胶砂比砂浆界面处平均弹性模量分布图,可以更直观地看出随着胶砂比的减小,界面处的弹性模量逐渐减小。弹性模量由胶砂比为1∶1时的41.50 GPa,减小至胶砂比1∶2的33.89 GPa、胶砂比1∶3的30.77 GPa。这可能是随胶砂比减小,砂浆试块力学性能降低的原因。

图10 (a)胶砂比1∶1砂浆界面处弹性模量云图和(b)胶砂比1∶1砂浆界面处平均弹性模量Fig 10 (a) Contour maps of elastic modulus at the interface of mortar with 1∶1 cement-sand ratio and (b) average elastic modulus at the interface of mortar with 1∶1 cement-sand ratio
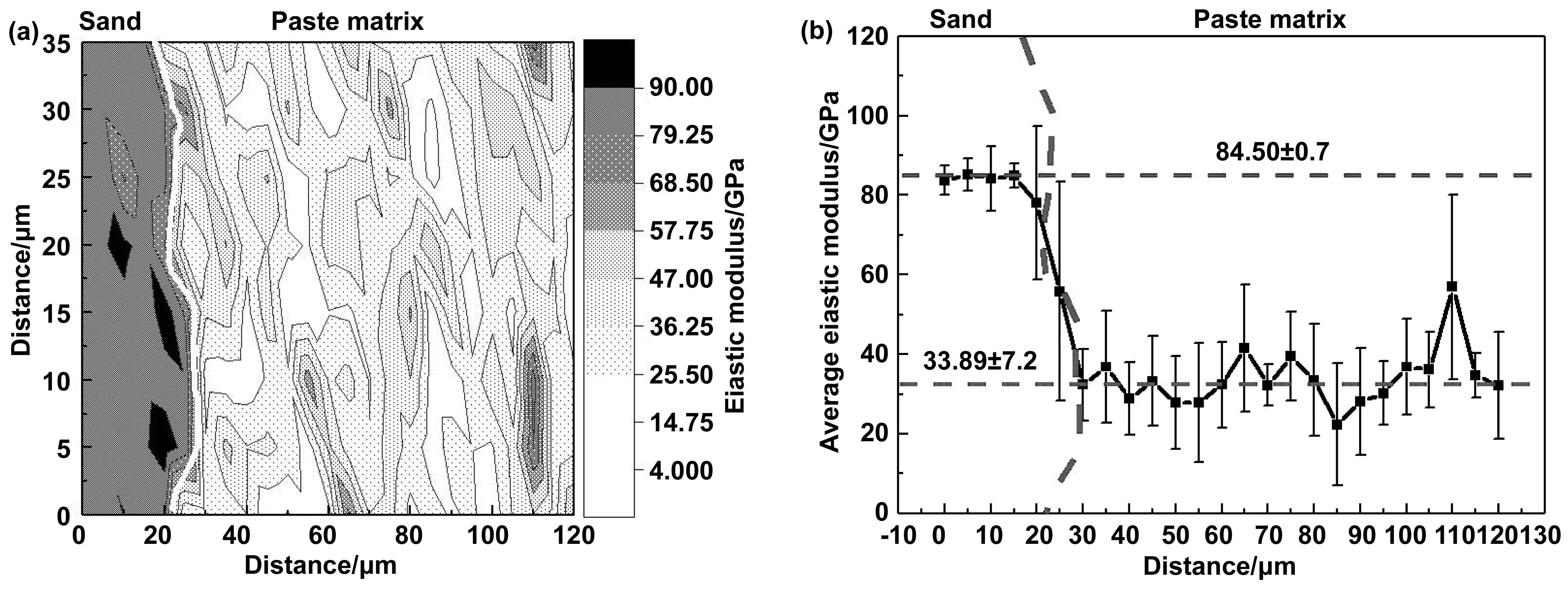
图11 (a)胶砂比1∶2砂浆界面处弹性模量云图和(b)胶砂比1∶2砂浆界面处平均弹性模量Fig 11 (a) Contour maps of elastic modulus at the interface of mortar with 1∶2 cement-sand ratio and (b) average elastic modulus at the interface of mortar with 1∶2 cement-sand ratio
2.6 界面的微观形貌
不同胶砂比试块碳化后微观形貌如图13所示,从图中可以清晰的看到砂浆界面由砂子,砂浆界面过渡区、胶凝材料基体3部分组成。当胶砂比为1∶1时,砂子和浆体粘结的非常紧密,界面结构致密,界面处没有孔隙和微裂缝。随着胶砂比的逐渐降低,界面结构变得疏松,出现微裂缝,微裂缝的宽度逐渐增大。胶砂比为1∶3时,砂子和浆体出现层错。随着砂子掺量的增加,界面结构变得越来越差,这与纳米压痕和孔结构结果是一致的。胶砂比1∶1试块碳化后砂子界面处的扫描电镜图如图14所示。通过增大图像的放大倍数可以看到,图中出现了许多形状不规则且表面光滑的晶体。对这些晶体进行点扫描分析,发现它们均是碳化产物CaCO3。说明砂浆试块碳化之后,碳化产物CaCO3会在砂子表面富集[24]。随胶砂比的减小,砂浆试块的砂子掺量逐渐增多,砂浆界面也随之增多[25],碳化产物CaCO3在砂子表面的富集使胶凝材料基体与砂子的粘结性变差。这可能是砂浆试块力学性能随胶砂比减小而降低的原因。

图13 不同胶砂比试块碳化后微观形貌:(a)胶砂比1∶1;(b)胶砂比1∶2;(c)胶砂比1∶3Fig 13 Micromorphology of samples with different cement-sand ratio after carbonation

图14 胶砂比1∶1试块碳化后砂浆界面处扫描电镜图Fig 14 SEM images and EDS of the carbonated paste adjacent to siliceous river sand
3 结 论
(1) 用工业原料石灰石和砂岩在1 275 ℃制备了一种以C2S、C3S2为主、C2AS为辅的低钙固碳胶凝材料。其中β-C2S、γ-C2S、C3S2、C2AS的质量分数分别为33.2%、25.4%、25.8%、15.6%。
(2) 低钙固碳胶凝材料和硅质河砂拌合制成砂浆。随胶砂比的减小,抗压强度先增大后减小,当胶砂比为1∶1时抗压强度达到最大,其值为46.9 MPa。
(3) 当胶砂比为1∶1时,掺入的砂子较少,不会影响试块整体的胶凝性,反而河砂在低钙固碳胶凝材料砂浆碳化硬化过程中可以起到稀释的作用,有利于碳化反应的进行,使其碳化后的抗压强度高于净浆。
(4) 进一步降低胶砂比,砂浆试块孔隙率逐渐变大、界面处孔隙率变大且结构变差以及CaCO3会在砂浆界面处富集,这些是砂浆试块力学性能随胶砂比减小而降低的原因。
