基于UPLC-MS法快速定量检测人血浆中短链脂肪酸
2022-05-08赵海燕黄婷婷陆金玲陈宇昕王少兵
赵海燕,黄婷婷,陆金玲,陈宇昕,王少兵
(中南民族大学 药学院,武汉 430074)
短链脂肪酸(short chain fatty acids,SCFAs)是一类碳原子数小于6的羧酸类化合物[1].直链的SCFAs是由食物中的膳食纤维、糖醇等在结肠内被肠道微生物发酵而成的小分子代谢产物,其中乙酸、丙酸和丁酸在肠道内的含量占总SCFAs含量的90%[2].当SCFAs在肠道内产生后,除了通过粪便排泄外,还能迅速进入全身循环系统,对调控机体正常生理功能起着非常重要的作用[1].据报道,SCFAs中的丙酸和丁酸是重要的信号传导分子,能与G蛋白偶联受体结合,抑制组蛋白去乙酰化酶的活性[3].SCFAs与糖尿病[4]、炎性肠病[5]、结肠癌[6]、阿尔茨海默症[7-8]等有着密切的关联.因此,准确、全面地测定SCFAs的含量,对于疾病的诊断与治疗,有着非常重要的意义.
SCFAs结构简单,但直链与支链的化合物极性相似,且不同碳原子数的酸在体内的含量差异较大,因此要全面地测定各SCFAs的含量非常棘手.SCFAs挥发性强,多采用气质联用技术(GC-MS)测定粪便中SCFAs的含量[9-11].但SCFAs对热不稳定,在高温条件下样品损失多,采用GC法直接检测样品时,灵敏度不高.常采用五氟苄基溴、三甲基硅烷、氯甲酸苄酯等试剂,将SCFAs衍生化后,再用GC检测,以提高检测的灵敏度[12-14].但是这些衍生化反应或所需时间长至2 h;或反应条件苛刻,要求无水环境;或需要加入DMSO来进行乳化,操作过程繁琐,使GC-MS在应用上受到很大的限制.近年来,LC-MS也常被应用于SCFAs的含量测定.SCFAs含有羧基,可在负离子模式下,用LC-MS/MS检测,但在电喷雾离子源下,离子化效率低,直接检测的灵敏度不能满足生物基质中样品检测的需求[15].因此,常用带氨基的衍生化试剂衍生化后,在正离子模式下进行检测.CHAN等[16]采用12C和13C标记的苯胺衍生化方法与UPLC-MS/MS结合,测定了人粪便中12个SCFAs的含量.该衍生化方法在4℃下进行,反应条件温和,但需要反应2 h并加入反应终止剂.ZENG等[17]将待分析物与苄氧胺盐酸盐(O-BHA)在25℃下反应1 h后,萃取衍生化产物,再用UPLC-MS检测粪便中的12个SCFAs及酮体,操作过程略微繁琐.LIEBISCH等[18]用3-NPH衍生化方法与液质联用建立了人粪便中的4个SCFAs含量检测的方法.FU等[19]采用4-氨甲基喹啉衍生化方法成功地检测了人粪便中的7个SCFAs.上述衍生化方法反应条件易控制,操作方便,检测灵敏度较GC-MS法要高出近百倍,检测限可达到ng甚至pg级别,但检测对象均不包括2-甲基戊酸及3-甲基戊酸.此外,同位素标记的内标物,可以减少质谱中共流出物的干扰,近年来,此法也广泛应用于SCFAs这种结构差异小的化合物的检测[20].
研究表明:SCFAs除了广泛分布于粪便及肠内容物中,还存在于血液、脏器组织等.以往的研究多集中于分析结肠或粪便中的SCFAs的含量,较少关注血浆中SCFAs的水平.由于SCFAs在血浆中的含量远低于粪便,故对分析方法有更高的要求.而从C2到C6,包括所有直链与支链异构体的11种化合物同时检测的方法尚未见报道.因此,本研究建立了一种基于3-NPH衍生化方法结合UPLC-MS/MS法,采用同位素标记的内标物,检测人体血浆中短链脂肪酸的含量,将11种SCFAs基本实现基线分离,准确测定出血浆中的SCFAs的含量,为临床上某些代谢性疾病的诊断与治疗提供了有力的支持.
1 实验部分
1.1 仪器与材料
高效液相色谱仪(Waters Acquity UPLC system);质谱仪(Xevo TQ MS,美国Waters);超声波清洗器(PS-80,深圳市深华泰超声洗净设备有限公司);电子天平(CP214,上海奥豪斯仪器);液氮(99.999%,武汉分析仪器).
正常人体血浆(上海冠东生物科技);色谱纯甲醇、乙腈、吡啶、甲酸(武汉弗顿试剂);3-硝基苯肼(3-NPH)、N-(3-二甲基氨基丙基)-N′-乙基碳二亚胺盐酸盐(EDC),乙酸(C2)、丙酸(C3)、异丁酸(Iso-C4)、丁酸(C4)、2-甲基丁酸(2Me_C4)、异戊酸(3Me_C4)、戊酸(C5)、2-甲基戊酸(2Me_C5)、3-甲基戊酸(3Me_C5)、4-甲基戊酸(4Me_C5)、己酸(C6),同位素标记的内标物包括乙酸-d3(C2-2,2,2,-d3)、丙酸-d5(C3-d5)、丁酸-d7(C4-d7)、戊酸(C5-d9)、己酸(C6-d11),均购自于美国Sigma-Aldrich公司.
1.2 色谱条件
色谱柱为Waters UPLC BEH C18柱(100 mm×2.1 mm I.D.,1.7µm),柱温40℃,样品室温度为10℃,进样量5µL.流动相A为超纯水,B相为乙腈-甲醇(V(乙腈)∶V(甲醇)=2∶1),按照表1进行梯度洗脱.
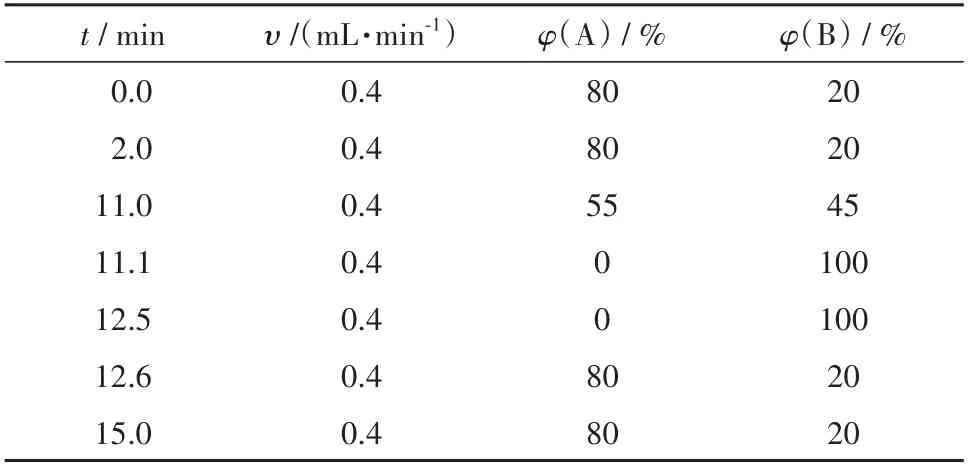
表1 液相检测梯度洗脱条件Tab.1 Gradient elution conditions for UPLC
质谱检测采用ESI电喷雾离子源,以正离子多反应监测离子模式检测,具体质谱参数为:离子喷雾电压5500 V,离子源温度550℃,去簇电压(DP)60 V,入口电势(EP)10 V,碰撞室出口电压(EP)10 V.
1.3 样品的制备
1.3.1 对照品贮备液的配制
精密称取SCFAs对照品各10.0 mg,置于2 mL容量瓶中,用水稀释定容配制成浓度为5.0 mg·mL-1的贮备液.精密称取同位素标记的内标标准品13C2-d3、C3-d5、C4-d7、C5-d9、C6-d11各10.0 mg,分别置于2 mL容量瓶中,用水稀释定容,即得浓度为5.0 mg·mL-1的内标贮备液,于-20℃下保存.
1.3.2 线性工作溶液的配制
内标物混合液:适量量取5种内标物贮备液,混合,用甲醇稀释定容至13C2-d3为20µg·mL-1,其他4种内标物为2µg·mL-1的内标物混合液,于4℃下保存.
标准品混合液:量取适量11种标准品的贮备液,混合,用甲醇序列稀释得到9个不同浓度的标准品混合液,其中乙酸的浓度分别为50、100、200、500、1000、2000、5000、10000、20000、50000 ng·mL-1,其余10种标准品的浓度分别为5、10、20、50、100、200、500、1000、2000 ng·mL-1.
1.4 血浆样品的制备
精密吸取60µL的血浆于1.5 mL的离心管中,并加入12µL内标物混合液和288µL甲醇,涡旋混合1 min后,于4℃下,13000 r·min-1离心10 min;吸取270µL上清液,依次加入50µL 200 mmol·L-1的3-NPH,12µL含0.6%吡啶溶液的1 mol·L-1的EDC,最后加入甲醇直至体系总体积为400µL,于40℃下孵育30 min,迅速取出,供色谱分析.
1.5 色谱方法学验证
1.5.1 线性和范围
标准溶液的校准曲线:取45µL标准品混合液,加45µL纯水稀释,加180µL甲醇、9µL内标物混合液,按照“1.4”项下加入NPH和EDC-吡啶溶液进行衍生化反应,供LC-MS/MS分析.
血浆加标溶液的校准曲线:取60µL血浆,加入等体积的标准品混合液、12µL内标混合液及228µL甲醇,按照“1.4”项同法操作,进行LC-MS/MS分析.
以色谱峰峰面积(分析物/内标物)的比值为y轴变量,药物浓度作为x轴变量,进行线性分析,通过加权(1/x2)最小二乘法拟合校准曲线,定量下限(LLOQ)在信噪比为10的条件下获得.
1.5.2 精密度
分别精密吸取5µL的低、中、高三个浓度的质控标准品溶液,按“1.2”项下液质条件测定SCFAs的含量,分析3次,计算RSD值,考察仪器的精密度.
1.5.3 基质效应与回收率
取适量的血浆样品,以甲醇沉淀蛋白,加入一定量的标准品溶液和内标溶液,对样品进行衍生化反应,测得的峰面积记为Aafter.取血浆样品,加入适量的标准品溶液和内标溶液,摇匀,甲醇沉淀蛋白后,按上述方法进行反应检测,测得样品的峰面积记为Abefore,提取回收率即为(Abefore/Aafter)×100%.
在纯溶剂中加入各标准品溶液,按溶液中标准曲线的样品的制备方法操作,测得的衍生化产物的峰面积记为A溶液.基质效应即为(Aafter/A溶液)×100%.
1.5.4 稳定性
将含血浆基质的待测标准品溶液按照“1.4”项下进行衍生化反应,于48 h内多次进样分析,考察样品的稳定性.
2 结果与讨论
2.1 色谱条件的优化
该反应选择多反应检测模式(MRM),通过对比发现正离子模式下检测的离子强度是负离子模式下的10倍,因此选择正离子模式监测.优化后的各参数结果见表2.在色谱分离方面,尝试了甲醇-水、乙腈-水两种混合溶剂系统,结果发现:以乙腈为流动相时,色谱峰尖而窄,但2Me_C5与3Me_C5基本重叠;以甲醇为流动相时,11个SCFAs都能分离开,但色谱峰矮而宽.随后,以甲醇与乙腈混合溶液作为有机相,发现当乙腈和甲醇体积比为2∶1时,得到峰形尖锐且对称、分离度较好的色谱图,结果见图1.

图1 SCFAs检测的UPLC-MS色谱图Fig.1 UPLC-MS chromatograms for derivatives of SCFAs
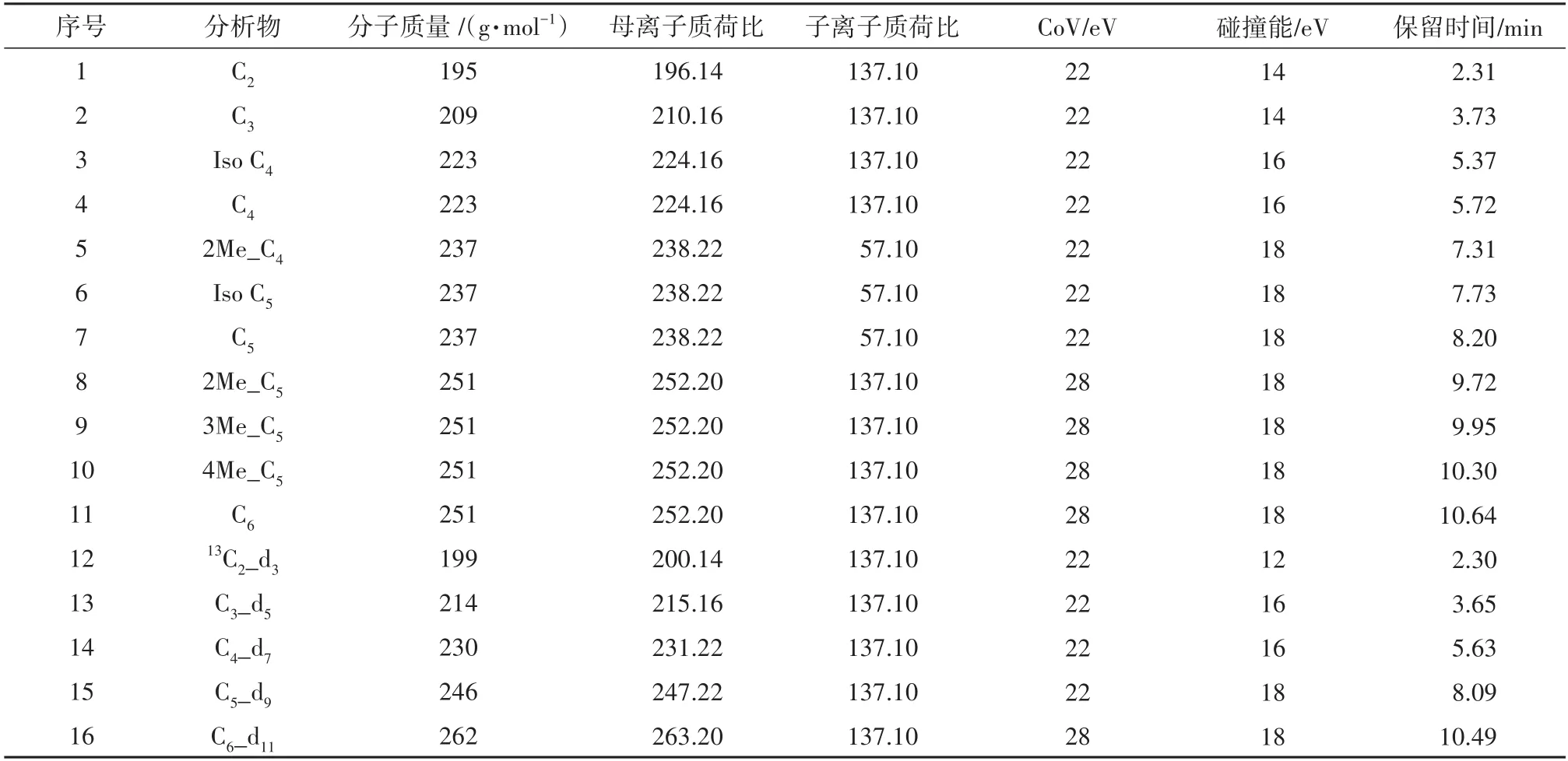
表2 SCFAs衍生化产物的优化MRM检测条件Tab.2 Optimum MRM detection conditions for derivatives of SCFAs
2.2 衍生化条件的优化
首先,考察了3-NPH的浓度与EDC在反应体系的终浓度对衍生化产物的峰面积的影响,结果见图2和图3.由图2(a)可知:当EDC的浓度固定不变,3-NPH的终浓度由2.5 mmol·L-1增至25 mmol·L-1时,衍生化产物的峰面积显著增加;当浓度继续增加时,产物的峰面积不再增加,因此3-NPH的浓度确定为25 mmol·L-1.由图3可知:当EDC浓度不变,增加3-NPH的浓度时,产物峰面积反而略微下降,说明增加3-NPH的浓度并不利于反应的进行;当3-NPH的浓度不变,EDC的浓度从15 mmol·L-1增至25 mmol·L-1时,各产物的峰面积明显增大;EDC的浓度继续增大至30 mmol·L-1时,C5及C6的峰面积显著增加,而对其他SCFAs并未有明显变化.综合考虑,确定衍生化试剂3-NPH的终浓度为25 mmol·L-1,EDC的终浓度为30 mmol·L-1.

图3 3-NPH和EDC浓度对SCFAs的衍生化反应的影响Fig.3 Effect of concentration of 3-NPH and EDC on the derivatization reaction of SCFAs
然后,考察了EDC中吡啶的含量、反应温度与反应时间对衍生化产物峰面积的影响,结果见图2.由图2(b)可知:将吡啶加入反应体系后,得到的产物峰面积至少增大3倍,但继续增大吡啶的比例,产物的峰面积不再增加.考虑到血浆中基质复杂,结合文献综合考虑,选择6%作为吡啶的比例.由图2(c)可知:随温度的升高,衍生化产物的峰面积先是迅速增加,再逐渐缓慢上升,当温度高于40℃时,峰面积无明显增加,因此,反应温度定在40℃.由图2(d)可知:当反应时间超过20 min后,产物的峰面积增加缓慢,故将反应时间定在30 min.
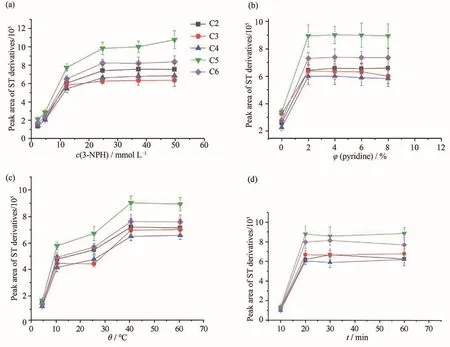
图2 反应条件对反应产物峰面积的影响Fig.2 Effect of reaction conditions on the the peak areas of derivatives
2.3 方法学考察
2.3.1 线性和定量下限
由于正常人体体内也存在痕量的SCFAs,难以获得完全不含SCFAs的空白血浆,本文分别制备了在溶液中及在血浆中进行反应的线性曲线,以SCFAs的浓度为横坐标,SCFAs峰面积与内标物的峰面积的比值为纵坐标,采用1/x2进行加权回归,绘制标准曲线,并计算回归方程及相关系数,结果见表3和表4.在溶液中反应的标准曲线与在血浆中反应的标准曲线的斜率基本一样,因此可以将血浆中测得的分析物的峰面积代入在溶液中进行反应的标准曲线中来求得在血浆中的分析物的浓度.
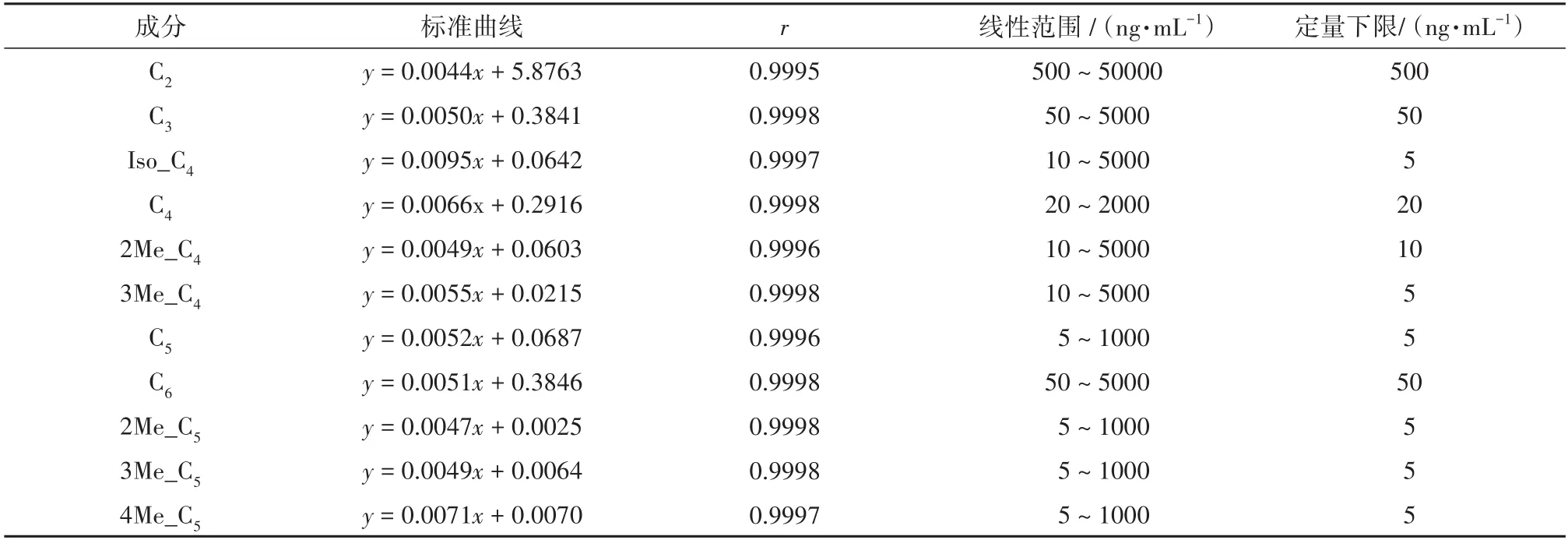
表3 在溶液中反应的SCFAs的标准曲线Tab.3 Linearity and range of SCFAs in solution
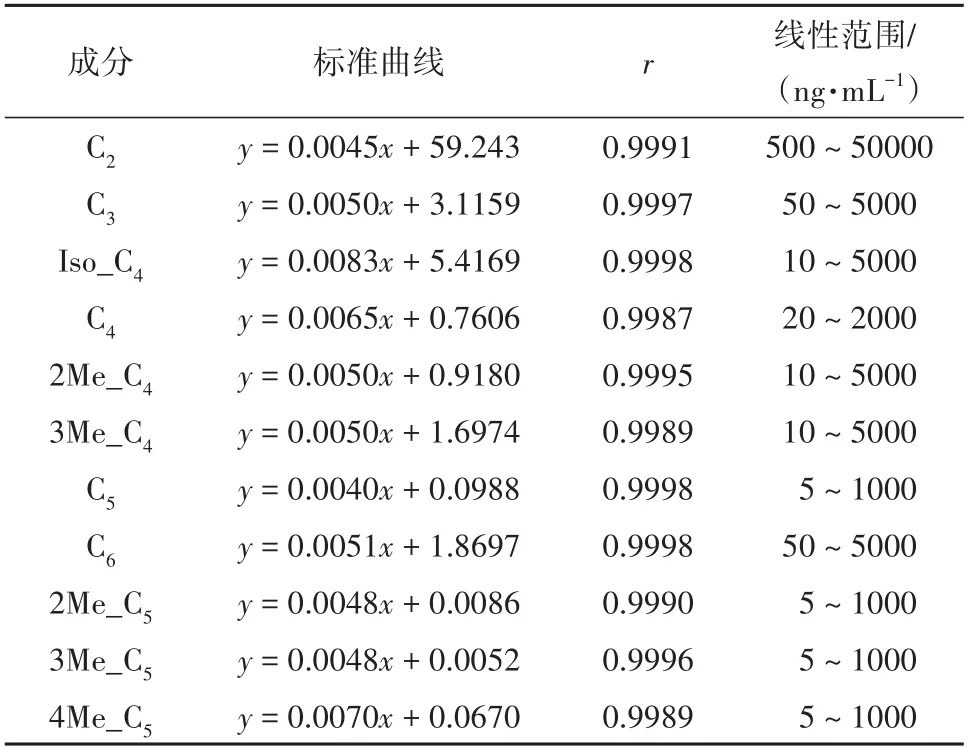
表4 在血浆中反应的SCFAs的标准曲线Tab.4 Linearity and range of SCFAs in plasma
2.3.2 精密度和准确度
采用低、中、高3个浓度分析评价各质控标准品溶液在血浆中的稳定性,按精密度试验方法操作,分别测定各SCFAs的峰面积的RSD值,结果见表5.表5中RSD均小于15%,表明该方法的精密度良好.
2.3.3 基质效应和提取回收率
基质效应和提取回收率的结果见表5,表5中基质效应在90%~115%之间,样品的平均回收率均在80%~120%之间,符合生物样品分析的要求.
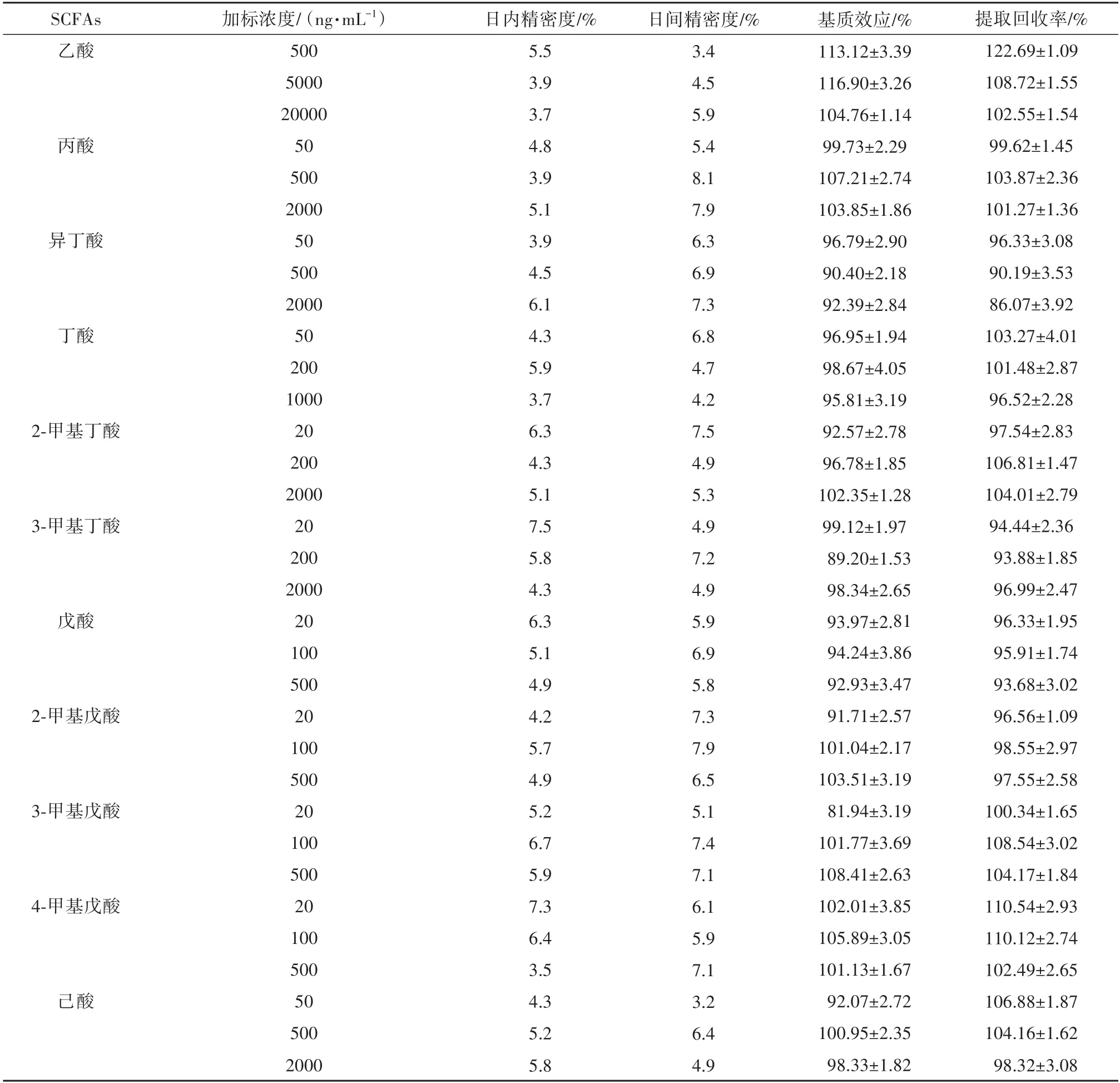
表5 精密度、基质效应、回收率试验结果(n=3)Tab.5 Results of precision,matrix effect and recovery test(n=3)
2.3.4 稳定性
取同一血浆样品,制备供试品溶液,考察3-NPH衍生的SCFAs在自动进样器样品托盘中放置48 h内的稳定性,结果见图4,结果表明样品在48 h内稳定性良好.

图4 SCFAs衍生化产物的稳定性结果Fig.4 Stabilities results of SCFAs derivatives
2.4 血浆的定量
正常血浆中SCFAs的含量测定结果见表6.表6中除了3Me_C5无法检测出来外,另外10种SCFAs均能被检测出来,其中乙酸、丙酸及支链丁酸的含量高于其他SCFAs的含量,戊酸与己酸的含量较其余SCFAs的含量明显较低.
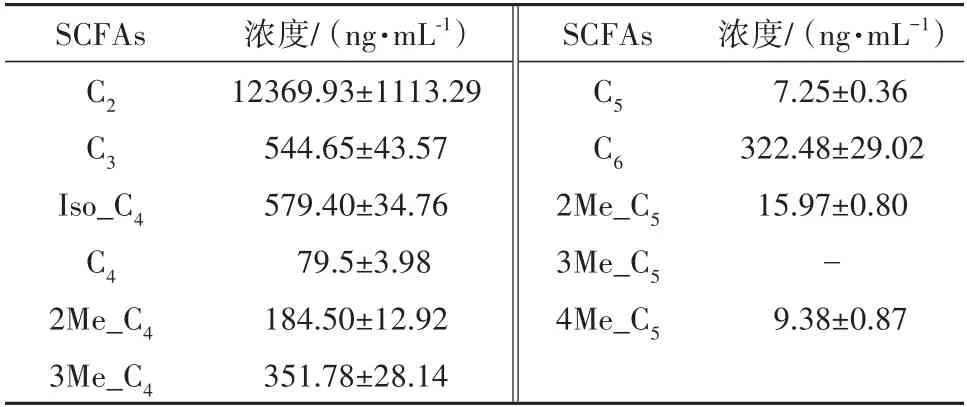
表6 正常血浆中SCFAs的含量测定结果(n=3)Tab.6 Determination of SCFAs in healthy human plasma(n=3)
3 结语
本文建立了基于3-NPH衍生化反应的UPLCMS/MS法以测定人体血浆中11种SCFAs的含量.建立的衍生化方法,反应条件温和易控制,反应时间适中.色谱方法学考察结果证明:该方法能在11 min内快速分离11种SCFAs,除3Me_C5无法检测外,每个峰都基本实现基线分离,各SCFAs回收率为80%~122%,日内、日间精密度<15%,SCFAs的峰面积与浓度有良好的线性关系(r>0.99).血浆样品在衍生化反应后可直接分析,不需要进行液液萃取、氮吹、复溶等步骤,该法操作简单易行,准确可靠,可应用于正常及病理血浆中SCFAs含量的测定,为临床多种疾病的诊断治疗提供依据.
