超高效液相色谱-串联质谱法快速测定鸡肉中氯羟吡啶药物的残留
2022-04-28邢骞文崔沙沙李文香高振同
邢骞文,崔沙沙,李文香,高振同
(河北省兽药饲料工作总站,河北 石家庄 050000)
球虫病是一种常见的寄生虫病, 寄生虫寄生于动物体内小肠部位并成倍增值, 进而导致动物组织损伤、采食量降低、养分吸收率下降,最终引起脱水和血液损失等严重损害。 氯羟吡啶是我国大量生产且使用最为广泛的抗球虫药物之一,由于其给药具有持续性,过量喂食易造成动物体细胞和生殖细胞的染色体、DNA 的畸变和损害[1]。 同时,聚积在动物体内的药物可能通过食物链进入人体,造成潜在危险与隐患。 我国在GB 31650-2019 《食品安全国家标准食品中兽药最大残留限量》 中明确规定鸡肉中氯羟吡啶的最高残留量为5000 微克/千克,但目前对于氯羟吡啶的检测方法非常有限, 关于鸡肉的相关报道更是屈指可数, 主要为高效液相色谱法、气相色谱-质谱法和液相色谱-串联质谱法[2-6]。 相较于前两种方法,液相色谱-串联质谱法的前处理时间短、有机试剂消耗量小,是分析氯羟吡啶残留较为理想的方法。 本文在超高效液相色谱-串联质谱法基础上,通过简易优化相关实验步骤,提高了实验效率、减少了有机试剂的消耗和污染, 为今后更加便捷快速地开展鸡肉中氯羟吡啶残留的检验检测工作提供了有力支撑和保证。
1 仪器和材料
1.1 试剂及材料
1.2 仪器
UPLC-XEVO TQ-S 超高效色谱仪-串联质谱联用仪(Waters 公司);BP211D 电子天平(赛多利斯公司);JJ200 电子天平(美国双杰兄弟);VXMTDG 涡旋振荡仪 (奥豪斯公司);KQ-500DV 超声波清洗器 (昆山市超声仪器有限公司 );HERAEUS BIOFUGE STRATOS CENTRIFUGE 离心机(Thermo 公司);N-EVAP112 氮吹仪(美国Jnc);MS3 digital 涡旋混合仪(IKA 公司);48PPM 半自动固相萃取仪 (美国J2 Scientific)
2 方法
2.1 色谱条件
色谱柱为HSS T3 柱 (2.1 毫米× 50 毫米,1.8 微米);流动相为0.1%甲酸水-乙腈,采用初始比例为0.1%甲酸水-乙腈(95∶5,V/V)的梯度洗脱进行目标药物的分离(见表1),流速为0.45毫升/分钟;柱温为35℃;进样体积为2 微升。

表1 液相梯度洗脱时间、流动相比例和曲线种类
2.2 质谱条件
串联质谱仪电喷雾离子源ESI+; 毛细管电压设定值为3.00 千伏; 脱溶剂气温度设定值为600℃;脱溶剂气流速设定值为1000 升/小时;碰撞气流速设定值为0.23 毫升/分钟。 多反应监测药物离子对及锥孔电压、碰撞能量见表2。

表2 氯羟吡啶的特征离子对、锥孔电压和碰撞能量
2.3 样品前处理
2.3.1 提取方法
称取均质好的鸡肉样品(2±0.02 克)于50 毫升离心管中,加入10 毫升80%乙腈水溶液(含0.2%甲酸),2500 转/分钟振荡10 分钟, 超声10分钟,10000 转/分钟离心10 分钟, 上清液作为备用液。
2.3.2 净化方法
准确移取3.00 毫升备用液过PRiME HLB柱,收集全部过柱液于10 毫升离心管内,氮气吹干,加入初始比例流动相0.60 毫升复溶。 涡旋1分钟, 超声使其充分溶解, 再次涡旋混合后过0.22 微米滤膜,供超高效液相色谱-串联质谱仪测定。
2.4 基质标的配制及标准曲线的绘制
称取适量氯羟吡啶标准物质,用甲醇稀释,配制成浓度约为200 微克/毫升的氯羟吡啶标准储备液,-20℃保存,有效期6 个月。 用相应的空白样品基质制备标准浓度系列,分别约为4、10、20、100、400、1000 纳克/毫升 (分别相当于测试样品中约含有4 微克/千克、10 微克/千克、20 微克/千克、100 微克/千克、400 微克/千克、1000 微克/千克的目标化合物),按2.1 和2.2 测定,以上机浓度为横坐标,以定量峰面积为纵坐标,进行线性回归计算,得出目标药物的线性方程和相关系数。
3 结果与讨论
3.1 色谱条件的选择
本实验分别选用0.005 毫升/升乙酸铵水溶液 (含0.05%甲酸)-甲醇、0.1%甲酸水-甲醇和0.1%甲酸水-乙腈作为流动相体系进行分析。 结果表明当选用0.1%甲酸水-乙腈为流动相时,目标药物色谱峰形更好, 响应更高, 故最终选用0.1%甲酸水-乙腈作为本实验流动相。 同时,实验测试了多种梯度洗脱方式,发现采用表1 的梯度洗脱对于消除杂峰干扰、分离目标药物的效果更佳,当流速设置为0.45 毫升/分钟时,分离效果最佳。
3.2 质谱条件的优化
实验发现在正离子模式下, 氯羟吡啶可产生较强的[M+H]离子峰,选择丰度最大的母离子192.0 质荷比及其对应的两个响应较强的子离子192.0>87.0 质荷比和192.0>101.0 质荷比作为氯羟吡啶的定性离子对,再从该离子对中选择响应值最高、基质干扰最小的子离子192.0>101.0 质荷比作为定量离子。 通过调谐,最终确定当锥孔电压为48 伏、碰撞能量分别为29 伏和24 伏时, 对应子离子的响应值最高。
3.3 样品前处理
3.3.1 提取液的选择
乙腈与甲醇相比, 可以更好地沉淀样品中蛋白质,为后续固相萃取时提供更加干净的提取液,提高整体净化效果[7]。 故本实验选用乙腈和80%乙腈水溶液(含0.2%甲酸)作为提取液进行试验, 结果发现 80%乙腈水溶液(含0.2%甲酸) 作为提取液时,氯羟吡啶特征离子色谱峰的面积更大且计算得出的回收率更高。 因为选用纯有机相作为提取液时,会让除目标药物之外的杂质通过固相萃取柱, 导致杂质干扰, 影响最终检测结果。
3.3.2 固相萃取柱的选择
现有文献中大多选用碱性氧化铝柱和silica 柱作为固相萃取柱, 本实验比较了碱性氧化铝柱、PRiME HLB柱对于药物的净化效果, 发现PRiME HLB 固相萃取柱不仅无需预洗、淋洗、洗脱等步骤, 并且对氯羟吡啶的萃取效果更好,目标药物的回收率更高。 同时,比较了该方法与SN/T 3144-2011 的前处理过程与实验结果,发现两者得出的标准曲线和相关系数近乎一致,但在处理效率、试剂耗材、节能环保等方面,本方法都有不可低估的优势。
3.4 标准曲线及检出限、定量限
配制4、10、20、100、400、1000 纳克/毫升的系列氯羟吡啶基质标准工作液,上机测定,以基质标准溶液浓度为横坐标,定量色谱峰面积为纵坐标绘制标准曲线 (图1), 回归方程为:y=2668.96x+5489.83,相关系数R2 为0.9923,满足残留定量分析要求,说明氯羟吡啶利用本方法在4~1000 纳克/毫升范围内呈现良好线性关系。 如图2 所示,特征离子在10 纳克/毫升时的信噪比满足S/N>10 要求且远远高于10,说明本方法相较于SN/T 3144-2011 具有更高的灵敏度, 可以通过本方法确定更精确的检测线、定量限。
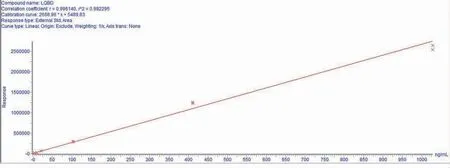
图1 不同浓度氯羟吡啶的线性关系

图2 特征离子在10ng/mL 时的信噪比
3.5 方法回收率和相对标准偏差
在前处理前向空白鸡肉中添加10、20、100、400 微克/千克的氯羟吡啶药物进行回收率实验,每个添加量进行6 次平行性实验,回收率及相对偏差见图3、表3。该方法的四个添加水平回收率位于68.0%~90.4%, 相对标准偏差均小于1.5%, 说明该方法的准确度和精密度均得到了很好的保证。

图3 鸡肉添加氯羟吡啶定量离子色谱图
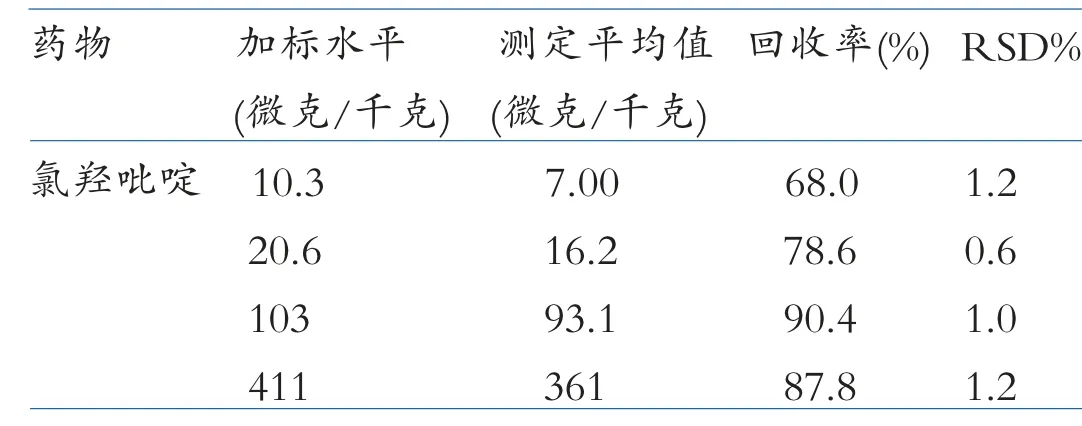
表3 鸡肉中氯羟吡啶的添加回收率的实验结果
4 结论
本文研究了超高效液相色谱-串联质谱法检测鸡肉中氯羟吡啶的方法,通过优化提取、 净化等步骤建立了可信度高、线性良好的快速检测方法,相较于已报道的方法, 具有明显的高效率、低耗材、更环保等优势,能够满足国内外对氯羟吡啶的检测方法与残留限量的要求,为今后大批量开展鸡肉中氯羟吡啶的验验检测工作提供了有力保障。