第一性原理研究合金元素对逆变奥氏体在Cu 沉淀上异质形核的影响*
2022-04-27吴迪杨永章小峰黄贞益王昭东
吴迪 杨永† 章小峰 黄贞益 王昭东
1) (安徽工业大学,冶金工程与资源综合利用安徽省重点实验室,马鞍山 243002)
2) (安徽工业大学冶金工程学院,马鞍山 243000)
3) (东北大学,轧制技术及连轧自动化国家重点实验室,沈阳 110819)
超高强度马氏体钢的力学性能强烈依赖于逆变奥氏体的形状、尺寸及含量.通常,提高逆变奥氏体含量,有助于改善超高强度钢的塑韧性.对含Cu 马氏体淬火钢时效处理时,Cu 粒子会在马氏体组织边界沉淀,并作为质点促进逆变奥氏体形核.为了探索不同合金元素对逆变奥氏体在Cu 沉淀上异质形核的影响,本文利用第一性原理方法研究了合金元素X (X=Cr,Al,Mo,W,Ni,Co,Mn)对Cu/γ-Fe 界面性质的影响,并分析了合金原子替换界面处Cu 和Fe 原子前后界面的黏附功、界面能和电子结构.研究结果表明,合金元素Cr,Mo,W,Mn 替换Cu/γ-Fe 界面处Cu 原子时,Cu/γ-Fe 界面处产生强烈的X—Fe 共价键,黏附功增大且界面能减小,显著提高界面稳定性,促进γ-Fe 在Cu 沉淀上异质形核.而替换界面处Fe 原子时,界面稳定性变化很小,掺杂原子与相邻的其他原子成键较弱.
1 引言
随着高尖端领域对钢材性能要求越来越高及轻量化需求上升,具有高强度高塑韧性的马氏体基超高强度钢广泛应用于航空航天、桥梁工程和军事尖端领域[1−4],如应用于发动机罩、火箭和导弹的外壳体、飞机起落架、机身骨架、高压容器组件、高强度螺栓、大跨度悬索桥的缆绳等.马氏体钢[5−7]具有超高强度的原因是多方面的,主要包括马氏体转变时在晶体内产生大量的微观缺陷(位错、孪晶及层错等)阻碍位错运动所产生的相变强化,马氏体形成后碳及合金原子向位错或其他晶体缺陷处扩散偏聚、形成析出相钉扎位错的时效强化以及固溶强化等,而其高韧性主要源于逆变/残余奥氏体对裂纹尖端的钝化及促进裂纹转向的作用.
马氏体组织内存在的大量位错及析出相是超高强度钢具有超高强度的基础[8],但也因此损害了马氏体钢的韧性.向马氏体钢中添加一定量的合金元素,获得的超高强度钢能够同时拥有良好的韧性.合金化超高强度钢之所以拥有如此优异的性能,是因为合金元素能够参与相变而影响奥氏体含量,从而改变材料的力学性能.大量研究[9,10]表明,想要同时拥有高强度高韧性,需要在马氏体钢中保留一定量的奥氏体.Ni 和Mn 是奥氏体形成元素,能够增加奥氏体稳定性,添加Ni 和Mn 元素使得超高强度钢中奥氏体含量增加,从而提高材料韧性.Tomohiko 等[11]向超高强度TBF 钢中复合添加Al-Nb-Mo,发现Al 元素促进大量的残余奥氏体稳定存在,而Nb 和Mo 元素的加入细化了晶粒,提高了TBF 钢的夏比冲击吸收值,使材料获得了优异的韧性.He 等[12]对13 Ni 马氏体时效钢掺杂合金元素做了大量实验,结果表明Ti 元素的添加使材料的强度很高,但韧性极差.而将Ni 元素含量控制在18%时,可有效提高材料的延展性和韧性.Machmeier 等[13]还发现,将Cr 元素含量从2%增加到3%时,超高强度钢中逆变奥氏体含量增加,虽然材料的强度有所下降,但韧性大幅提高.Gruber 等[14]研究马氏体钢回火时发现,沿着马氏体板条边界形成许多逆变奥氏体薄膜,这些逆变奥氏体薄膜能够钝化裂纹,显著提高材料塑韧性.Govindaraj 等[15]发现,马氏体钢时效时在马氏体板条边界会产生逆变奥氏体和Cu 沉淀,析出Cu粒子不仅强化组织,还作为逆变奥氏体的异质成核位点,促进富镍锰逆变奥氏体形核,进一步提高马氏体钢的塑韧性,得到了高强度高韧性的马氏体钢.Huang 等[16]发现,在时效初期,Cu 粒子中富含Ni,Mn 和Al 原子,随时效时间延长,Ni,Mn 和Al 原子逐渐从Cu 粒子中被排出而富集在Cu 粒子界面处,表明Cu 粒子中部分Cu 原子被合金原子取代.
截止目前,超高强度马氏体钢中不同合金化方式对时效过程中Cu 沉淀上奥氏体异质形核的影响尚不明确,因此,研究合金元素对Cu 沉淀上奥氏体异质形核的影响对提高马氏体钢韧性具有重要的理论及工程意义.基于此,建立了Cu/γ-Fe 界面模型,利用第一性原理,从原子尺度研究了合金元素X(X=Cr,Al,Mo,W,Ni,Co,Mn)对逆变奥氏体在Cu 沉淀上异质形核的影响,研究结果有助于揭示含Cu 超高强度钢中不同合金化方式的潜在影响.
2 计算方法
使用基于密度泛函理论结合平面波赝势的CASTEP 软件[17]进行计算,选用广义梯度近似(generalized gradient approximation,GGA)中的(Perdew-Burke-Ernzerhof) PBE 泛函[18]来表示交互作用和关联效应,采用超软赝势来描述原子核和价电子的相互作用.所有计算均在倒易空间上进行,通过布里渊区计算体系电荷密度及总能量,晶胞所采用的的K点网格为10 × 10 × 10,表面模型取10 × 10 × 1,Cu(010)/γ-Fe(010)界面模型取5 × 10 × 1.计算中最大平面波截断能设置为400 eV,收敛条件是自洽计算的最后两个循环能量之差小于1 × 10–6eV/atom,作用在每个原子上的力不大于0.03 eV/Å,内应力不大于0.05 GPa.
3 建立晶体模型
建立界面模型之前,需优化晶胞获得能量最低构型,使Cu 和γ-Fe 单晶胞原子坐标、晶格参数均达到稳定状态.完全几何优化后,Cu 的晶格参数为a=b=c=0.3629 nm,γ-Fe 的晶格参数为a=b=c=0.3435 nm.根据Bitter 和Kaufmann[19]的研究,时效析出的Cu 和γ-Fe 基体之间的位向关系为(010)[010]Cu//(010)[010]γ-Fe.基于此位向关系,分别建立Cu(010)面和γ-Fe(010)面的表面模型.
3.1 确定原子层数
研究Cu(010)/γ-Fe(010)界面性质,需要先对Cu(010)和γ-Fe(010)面表面能进行收敛测试,确定构建Cu(010)/γ-Fe(010)界面模型的原子层数,使远离界面处的原子呈现体相特征.随着表面原子数目的增加,对晶胞结构的描述越来越准确,但是消耗的计算时间和软件资源则会大大增加,因此选择1 × 1 单晶胞进行表面能收敛测试.Cu(010)和γ-Fe(010)表面均只含一种原子,属于极性表面,为了避免偶极矩效应的发生,采用奇数层原子[20].表面能计算公式为[21−23]
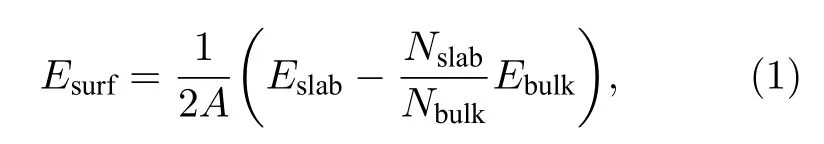
式中,Esurf表示所求体系的表面能(J·m–2);Eslab表示切层后添加真空层表面体系的总能量(eV);Ebulk表示单晶胞体系总能量(eV);Nslab和Nbulk分别表示添加真空层表面体系和单晶胞体系中原子个数;A为表面面积(nm2).表1 为Cu(010)和γ-Fe(010)表面能随原子层数增加而不断收敛的变化趋势.从表1 中可以看出,当Cu(010)和γ-Fe(010)原子层数N在7 层时,表面能分别收敛于1.44 J·m–2和3.75 J·m–2.因此,选取7 层的Cu(010)和7 层的γ-Fe(010)构建界面模型.
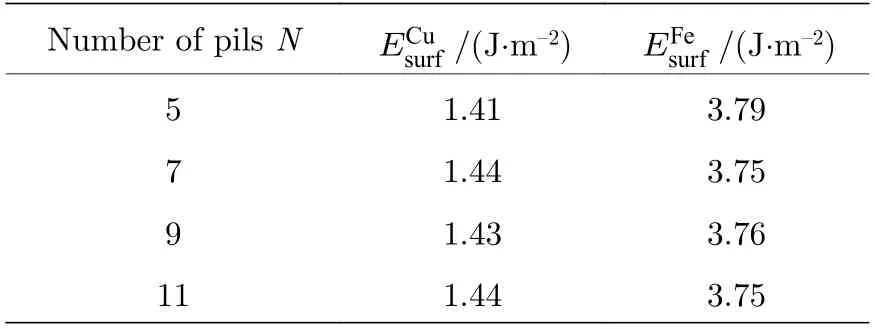
表1 Cu(010)和γ-Fe(010)表面能收敛趋势Table 1.Convergence of Cu(010) and γ-Fe(010)surface energy.
3.2 界面模型的构建
分别建立7 层的Cu(010)和γ-Fe(010)表面模型,并将γ-Fe(010)堆垛在Cu(010)晶面上,为了消除上下表面之间的相互作用在晶体表面添加15 Å的真空层,最后以2 × 1 × 1 形式扩胞建立界面模型,共28 个原子.结构模型如图1 所示,假设界面处Ⅰ和Ⅱ位置分别被合金X(X=Cr,Al,Mo,W,Ni,Co,Mn)替换一个Cu 或Fe 原子,界面模型一共有14 层原子,固定上下各4 层原子,保留中间6 层原子充分弛豫,图2 为Cr 原子替换一个Cu 原子后优化至平衡态的界面模型.
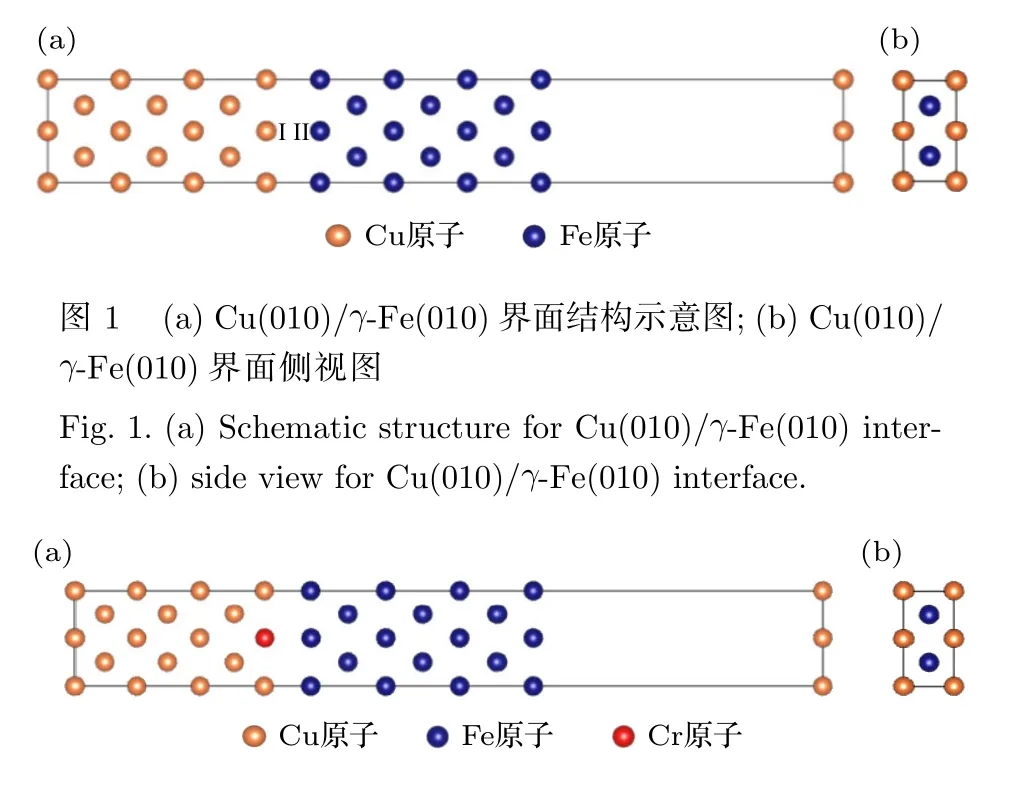
图2 (a) Cr 替换Cu 原子的Cu(010)/γ-Fe(010)界面结构示意图;(b) Cr 替换Cu 原子的Cu(010)/γ-Fe(010)界面侧视图Fig.2.(a) Schematic structure of Cr replacing Cu for Cu(010)/γ-Fe(010) interface;(b) side view of Cr replacing Cu for Cu(010)/γ-Fe(010) interface.
4 计算结果与讨论
4.1 黏附功和界面能
界面的稳定性和结合强度可以用黏附功表示,其定义为把一个界面分离成两个自由表面在单位面积上所做的可逆功.黏附功越大,表明界面原子之间相互的结合力越大,即破坏界面所需要的能量越多,界面越稳定.黏附功的计算公式为[24−27]
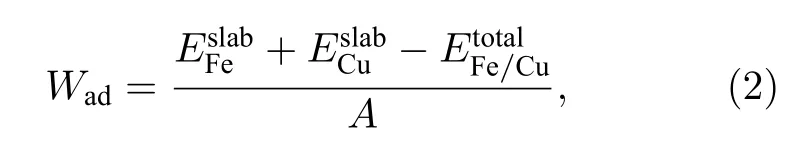
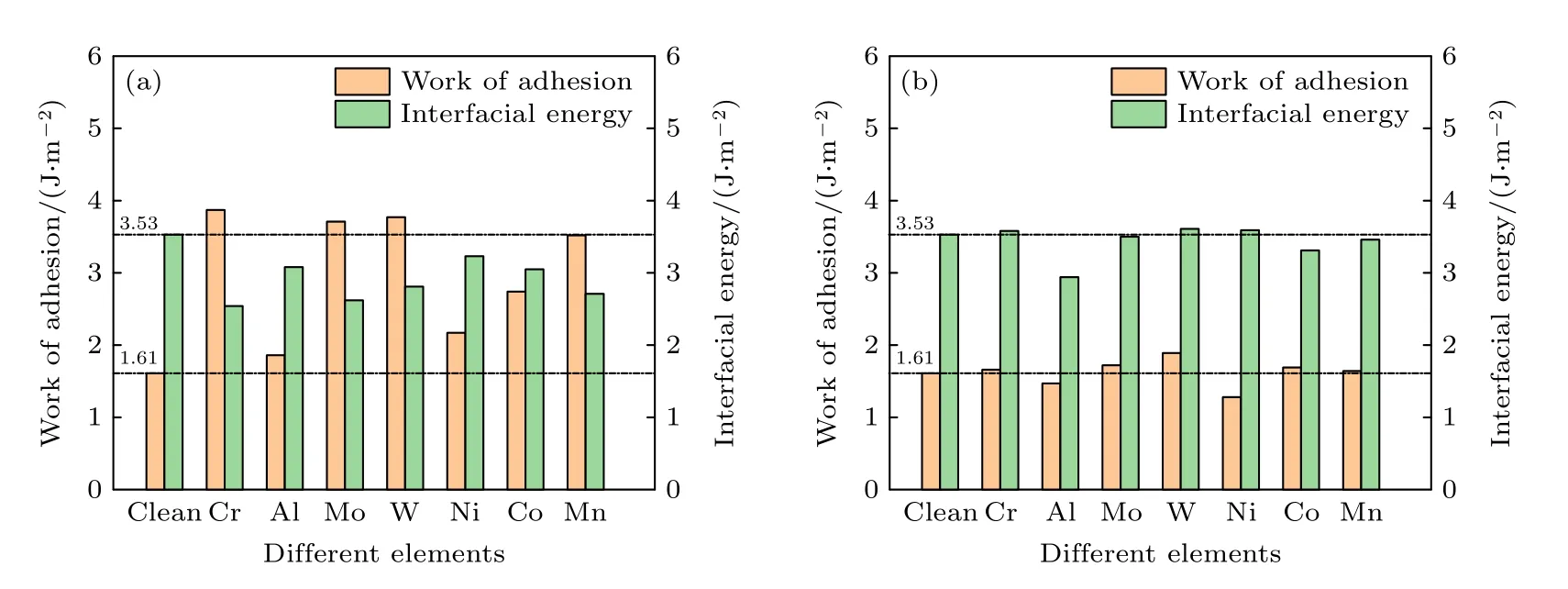
图3 (a) 合金元素替换Cu 后Cu(010)/γ-Fe(010)界面的黏附功与界面能;(b) 合金元素替换Fe 后Cu(010)/γ-Fe(010)界面的黏附功与界面能Fig.3.(a) Work of adhesion and interfacial energy for Cu(010)/γ-Fe(010) after replacing Cu atom;(b) work of adhesion and interfacial energy for Cu(010)/γ-Fe(010) after replacing Fe atom.
通过计算界面能可以进一步研究界面的稳定性,界面能越小,界面越稳定,越有利于Cu 析出成为逆变奥氏体异质形核的核心.Cu(010)/γ-Fe(010)的界面能可通过(3)式表示[28−31]:
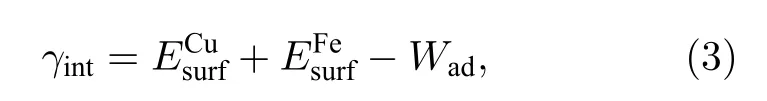
4.2 电子性质
为了进一步了解界面的键合情况,本文模拟计算了Cu(010)/γ-Fe(010)界面的电子结构,包括态密度、布居数、键长和电荷密度,从电子角度解释界面的稳定性.
4.2.1 分波态密度
图4 为合金原子替换Cu(010)/γ-Fe(010)界面处一个Cu 原子前后的分波态密度图,掺杂前的态密度波峰较平坦,而掺杂后局域尖峰增加,表现出较强的局域性.由图4(a)可知,掺杂前仅在电子态–3 eV 时Cu-d 轨道与Fe-d 轨道产生杂化,形成Cu—Fe 共价键,但键性较弱.掺杂Cr,Mo 和W 时,在电子态–7—0 eV 范围内合金原子的p,d 轨道和Cu,Fe 原子的p,d 产生强烈的电子轨道杂化,并伴随极强的电子峰,形成强共价键,其中Cr 原子与Cu,Fe 原子形成的共价键键性最强,Mo,W 原子掺杂形成的共价键稍弱之.Mn 原子掺杂后,在电子态–4.2 eV 时Mn-s 轨道与Fe-s,Cu-s轨道重叠,在–3,–1.3 和0 eV 电子态时Mn-d 轨道与Fe-d,Cu-d 轨道重叠产生明显杂化,形成较强的共价键.Ni,Co 原子掺杂后,在电子态–4.2 eV时Ni-s,Co-s 轨道与Fe-s,Cu-s 轨道重叠,形成X—Cu,X—Fe 共价键,但重叠峰数量相较于掺杂Cr,Mo,W,Mn 时要少得多,峰强度也较弱,因此形成的共价键更弱.Al 原子掺杂后,在电子态–3 eV 时Al-s 轨道与Fe-d,Cu-d 轨道均产生杂化,形成Al—Fe 共价键和微弱的Al—Cu 离子键.
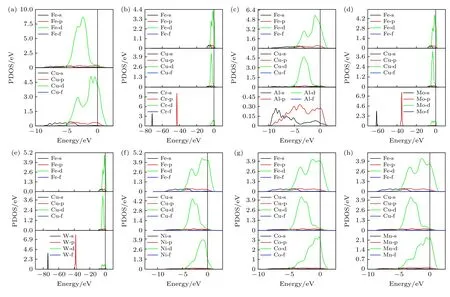
图4 (a) 掺杂前界面的分波态密度;(b)—(h) Cr,Al,Mo,W,Ni,Co 和Mn 替换Cu 原子后界面的分波态密度Fig.4.(a) Partial density of states on the interface for Cu(010)/γ-Fe(010) before doping;(b)–(h) partial density of states on the interface for Cu(010)/γ-Fe(010) after replacing Cu by Cr,Al,Mo,W,Ni,Co,and Mn,respectively.
图5 显示了Cu(010)/γ-Fe(010)界面处一个Fe 原子分别被不同合金原子替换的分波态密度图.对比图4 可以看出,Cr,Mo,W 替换Fe 原子和替换Cu 原子的分波态密度图十分相似,均在电子态–7—0 eV 范围内产生合金原子p,d 轨道和Cu,Fe 原子p,d 轨道的杂化,但替换Fe 原子时重叠峰明显减少,峰强度也较低,表明此时形成的共价键要弱于Cu 原子被替换时形成的共价键.Al,Ni,Co 原子替换Fe 原子后,分态密度图中合金原子和Fe 原子产生的轨道杂化很少,表明两原子间相互作用较弱.Mn 原子掺杂时,Mn-s 轨道与Cu-s,Fe-d 轨道在电子态–5.5 和–4 eV 处产生杂化,Mn-d轨道与Fe-d 轨道在电子态–3,–1.2 和–0.5 eV 处也产生杂化,形成的较强的共价键.

图5 (a)掺杂前界面的分波态密度;(b)—(h) Cr,Al,Mo,W,Ni,Co 和Mn 替换Fe 原子后界面的分波态密度Fig.5.(a) Partial density of states on the interface for Cu(010)/γ-Fe(010) before doping;(b)–(h) partial density of states on the interface for Cu(010)/γ-Fe(010) after replacing Fe by by Cr,Al,Mo,W,Ni,Co,and Mn,respectively.
综上,合金原子替换Cu 原子后产生了较多的轨道杂化,其中Cr 原子与相邻原子产生键性最强,Mo,W,Mn 形成的共价键较强,Co,Ni 形成的化学键要稍弱,掺杂Al 原子时键性较弱,但相较于未掺杂体系,掺杂后体系化学键的键性均显著加强;而替换Fe 原子后产生的杂化要少的多,只有Cr,Mn 掺杂体系的键性明显增强,表明Cu 掺杂后的界面稳定性要远强于Fe 掺杂界面,这也验证了前面黏附功和界面能的计算结果.
4.2.2 布居分析及键长
态密度定性地描述原子间的键合情况,而Mulliken 布居数通过计算原子间的电荷转移可以半定量地描述原子间的键合情况[32].一般来说,布居数越大,键合力越大,表明原子间的交互作用越强.键长同样是衡量原子间稳定性的重要参数,原子间成键越短,稳定性越高.
共价键是由共用电子对组成的,所以共用的电子对越多,形成的共价键越强.替换Cu 原子前后的电荷布居数结果列于表2,可以看出,界面处原子间电荷转移发生显著变化,Cr,Mo,Ni,Co,Mn失去电子转化为共用电子对的总电子数均高于未掺杂时的界面,提高了原子间的相互作用力,使掺杂体系的键性增强.Al,W 提供的共用电子对要少于掺杂前,但从表3 可以看出,掺杂后的键长明显缩短,如掺杂前Fe—Cu 键长为2.42 Å,掺杂后Al—Cu 键长为2.39 Å,W—Cu 键长为2.28 Å,其中W—Cu 键长远低于Fe—Cu 键,因此图3 计算结果显示,掺杂W 元素后黏附功增大.Cr 虽然转移的电荷为0.24,但形成的键长为2.06 Å,远短于其他合金原子的键长,因此Cr 掺杂的界面是最稳定的.
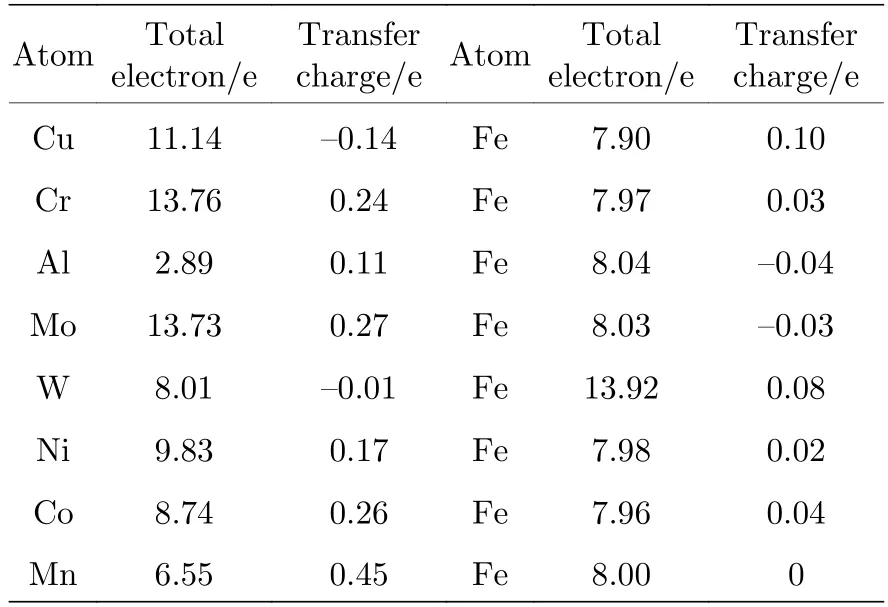
表2 合金元素X 替换Cu 前后界面布居分析Table 2.Mulliken population analysis on the interface for Cu(010)/γ-Fe(010) before and after replacement of Cu by alloying element X.
表3 合金元素X 替换Cu 和Fe 前后界面原子键长Table 3.Bond length on the interface for Cu(010)/γ-Fe(010) before and after replacement of Cu or Fe by alloying element X.
表4 是替换Fe 原子时的电荷布居数,转移的电荷较少,Cr,Mn 失去电荷分别为0.14 和0.33,高于未掺杂体系.其中Mn 原子失去的电荷为0.33,产生强大的吸引Cu 离子的正电场,形成较强的共价键.从图4(g)态密度图也同样得到,Mn原子与相邻原子间有较多的电子轨道杂化,这于前面计算的结果也是相符合的.而其他原子掺杂后没有明显提高键性,甚至使键长增加.键长与形成共价键的原子半径有关,原子半径越大,键长越长,键能越小.Cu 原子半径为145 pm,Fe 原子半径为126 pm,替换Fe 相较于替换Cu,半径较小的原子被替换时的形成的键能要低于替换半径原子较大的.
4.2.3 差分电荷密度
差分电荷密度可以清晰地得到成键电子耦合过程中的电荷移动及成键极化方向等性质.由前面的计算结果可知,当合金元素替换Cu(010)/γ-Fe(010)界面的Cu 原子时,界面的稳定性远高于替换Fe 原子时的界面,因此对Cu 掺杂后界面的差分电荷密度进行了计算,如图6 所示.图中红色表示电子缺失,蓝色表示电子富集.图6(a)为掺杂前的界面,可以看出相邻的Cu 原子都失去电荷,表现为带正电的电子核,形成电荷贫化区,使得Cu 原子间形成金属键.界面处Cu,Fe 原子均表现为失去电荷,但Fe 周围存在大量电荷,并且沿着Cu—Fe 方向富集,表明形成Cu—Fe 共价键.当界面处Cu 原子被替换后,从图6(b)—(g)可以看出,合金原子和Fe 原子之间的电荷密度增加,表明原子间相互作用加强.其中Cr,Mo,W,Mn 与Fe 形成极强的X—Fe 共价键,均显著提高界面稳定性.而合金原子与Cu 原子之间也存在电荷累积,沿着Cu-X方向,形成强共价键.Al 和Ni 原子与Fe,Cu 原子均存在少量电荷转移,形成弱离子键,因此掺杂Al,Ni 的界面为离子键和共价键共存,但以共价键为主.以Cr,Mo,W,Mn 元素替换Cu(010)/γ-Fe(010)界面处的Cu 原子进行材料成分设计,能够获得更稳定的界面.
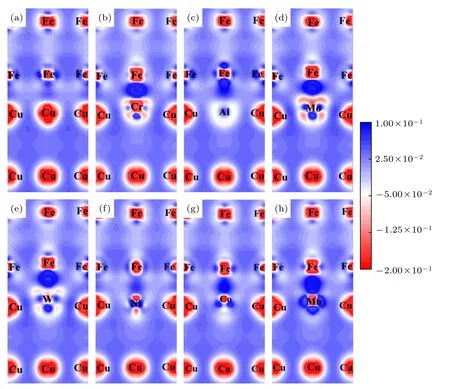
图6 替换Cu 原子前后Cu(010)/γ-Fe(010)界面差分电荷密度Fig.6.Difference charge density for Cu(010)/γ-Fe(010) interface before and after replacing Cu by different alloy atoms.
5 结论
本文利用第一性原理方法计算了不同合金元素分别掺杂Cu(010)/γ-Fe(010)界面Cu 和Fe 原子的结合强度、界面能及电子结构,探究了不同合金元素对Cu(010)/γ-Fe(010)界面性质的影响,得出结论如下.
1) Cr,Al,Mo,W,Ni,Co,Mn 替换Cu(010)/γ-Fe(010)界面Cu 原子时,界面的黏附功升高而界面能下降,显著提高界面稳定性,提高逆变奥氏体异质形核能力,使逆变奥氏体更容易在Cu 沉淀上形核,而Cr,W,Ni 原子替换Fe 原子时,界面能上升,提升奥氏体异质形核的难度,不利于逆变奥氏体的产生.
2) 替换Cu 原子后,Cr,Al,Mo,W,Ni,Co,Mn 原子与界面处Fe,Cu 原子电子轨道存在杂化,而替换Fe 原子时,很少轨道杂化,原子间相互作用较弱.
3) 掺杂前,Cu(010)/γ-Fe(010)界面处存在Cu—Fe 共价键.Cu 原子被Cr,Mo,W,Mn 合金原子替换时,界面处形成强X—Fe 共价键,且X—Fe 键共价性要远高于Cu—Fe 共价键.Al,Ni 掺杂时形成X—Fe 共价键的同时,还有较弱的离子键形成.
