红肉火龙果成熟茎的全长转录组测序分析及蔗糖代谢相关基因分离
2022-04-25郑乾明王小柯马玉华
郑乾明,王小柯,马玉华
(贵州省农业科学院果树科学研究所, 贵阳 550006)
【研究意义】红肉火龙果(Hylocereuspolyrhizus)是仙人掌科(Cactaceae)量天尺属(Hylocereus)多年生果树作物,其叶退化为短刺,肉质茎是主要的源器官。植株具有耐旱的生理特点,已成为贵州喀斯特石漠化地区的特色经济作物。红肉火龙果果实含有丰富的可溶性糖、有机酸、维生素C、类黄酮和甜菜苷色素等风味物质和营养成分,深受消费者喜爱[1-6]。果实中积累的风味物质和营养成分均来自于源器官的光合作用合成,蔗糖作为最主要的光合产物,经维管束韧皮部运输至果实中参与代谢和贮存[7]。研究红肉火龙果植株源器官(成熟茎)的转录组模式,分离光合作用等重要代谢途径的关键基因,有利于阐明红肉火龙果植株蔗糖合成的分子机制,对改良果实品质,提高果实商品价值具有重要意义。【前人研究进展】红肉火龙果是近年发展的新兴水果,目前尚无基因组序列发布。当前仅有基于Illumina平台的第二代转录组测序,对其果实或根开展系列测序和研究。例如,对红肉和白肉火龙果果实发育多个时期的转录组测序和比较,获得若干参与甜菜苷色素合成的基因[8],以及1个潜在的候选MYB转录因子[9];对红肉火龙果根系在盐胁迫下的转录组测序分析,获得一系列上调和下调的差异表达基因[10]。针对红肉火龙果成熟果实与成熟茎的转录组测序分析,获得79 658个Unigene,平均长度为690 bp,约44.11%的转录本在公共数据库中注释[11]。同时,还获得若干在成熟茎中表达,参与蔗糖代谢和转运的Unigene,但均未获得基因全长。因此,目前针对红肉火龙果的转录组测序数据较少,仅有的转录组数据均由Illumina平台的第二代测序获得,其序列长度较短,难以获得目标基因的全长序列。近年来,由Pacific Biosciences公司开发的PacBio测序是应用较为广泛的第3代测序平台。在此基础上发展起来的全长转录组测序,具有长读长,无PCR扩增,无需拼接,较高的随机错误率等特点[12]。目前全长转录组测序主要用于构建全长转录组数据库、转录本可变剪接分析、转录本结构研究和完善基因注释等[13]。【本研究切入点】提取红肉火龙果成熟茎的总RNA,进行基于PacBio Sequel平台的全长转录组测序,获得全长转录组数据库。【拟解决的关键问题】获得红肉火龙果成熟茎的全长转录组序列,根据功能注释和分类结果筛选参与蔗糖合成和降解等重要代谢途径的候选功能基因,为后续开展基因功能研究奠定基础。
1 材料与方法
1.1 材料
于2018年11—12月,在贵州省镇宁县的火龙果种植园,选择红肉火龙果品种“紫红龙”。在成年结果植株上选取生长良好、无病虫害的健壮成熟茎,剪下后立即置于冰盒带回实验室。用无水乙醇擦净后晾干,然后切取茎组织液氮中速冻,置于-80 ℃保存备用。
1.2 核酸提取
利用Trizol试剂提取成熟茎组织的总RNA,使用琼脂糖凝胶电泳、紫外光分光光度计和Bioanalyszer 2100(Agilent Technologies)检测其质量与浓度。
1.3 全长转录组测序
后续文库构建、测序和数据分析委托安诺优达基因科技有限公司开展。首先富集含有Poly A的mRNA,反转录合成第一链cDNA,然后PCR扩增合成cDNA。构建SMRTbell文库并进行损伤修复和末端修复,片段两端连接测序接头,形成具有茎环结构的插入片段测序文库。利用PacBio Sequel(Pacific Biosciences)测序仪测序,获得原始数据。
原始数据过滤获得Polymerase reads,去除接头序列后获得Subreads并进行质控,获得高质量的插入片段序列。对来源于同一条Polymerase read的Subreads片段进行合并和纠错,获得环状一致性序列(Circular Consensus Sequencing,CCS)。分析CCS序列,根据是否含有完整接头和poly A序列筛选获得全长非嵌合体(Full-length no chimera,FLNC)序列。使用ICA(isoform-level clustering algorithm)算法,对FLNC进行相同结构间的自我聚类和纠错,产生去冗余Consensus序列。将未测完整的插入片段序列比对到Consensus序列进行纠错,获得准确度大于99%的高质量转录本。使用Blastn和Blastx程序在NCBI非冗余核酸和蛋白质数据库进行检索注释。
1.4 序列分析
利用Trans Decoder Release v3.0.1鉴定转录本序列中的编码序列(Coding sequence,CDS)。对获得的高质量转录本序列,在NCBI核酸数据库(Nucleotide Sequence Database,NT)、NCBI非冗余蛋白数据库(Non-Redundant Protein Database,NR)、蛋白质真核直系同源簇数据库(Eukaryotic Orthologous Groups,KOG)、基因本体论数据库(Gene Ontology,GO)、蛋白质家族域数据库(Protein Families Database,Pfam)、KEGG(Kyoto Encyclopedia of Genes and Genomes)数据库进行基因功能注释。通过与植物转录因子数据库(Plant Transcription Factor Database,Plant TFDB)比对查找转录因子。序列一致性计算使用DNAStar软件的MegAlign程序,序列拼接使用BioEdit软件。
2 结果与分析
2.1 全长转录组测序
从表1看出,通过PacBio Sequel测序,获得聚合酶Reads数量740 069条,碱基数量为15.8 Gb。聚合酶Reads平均长度为21 366 bp,N50长度为42 865 bp。过滤后获得插入片段(Subreads)数量共8 483 057条,碱基数据量为15.1 Gb。其平均长度为1775 bp,N50长度为2200 bp。将来源于同一环状分子的Subreads聚类,获得环状一致序列(CCS)序列共计481 124条,其碱基数据量为1.1 Gb。CCS平均长度为2378 bp,N50长度为2623 bp,平均测序次数为13.87次。

表1 红肉火龙果成熟茎的全长转录组测序结果
进一步筛选获得全长非嵌合体(FLNC)数量为333 226条,碱基数据量为0.7 Gb,平均长度为2071 bp,N50长度为2250 bp。将来源于同一转录本的FLNC聚类获得Consensus序列,并将非全长序列对Consensus序列校正和去冗余,获得转录本30 973条。大部分转录本长度分布于1.8 kb,平均长度为1846 bp(图1)。其中准确度大于99%的高质量转录本数量为30 313条,平均长度为1829 bp。
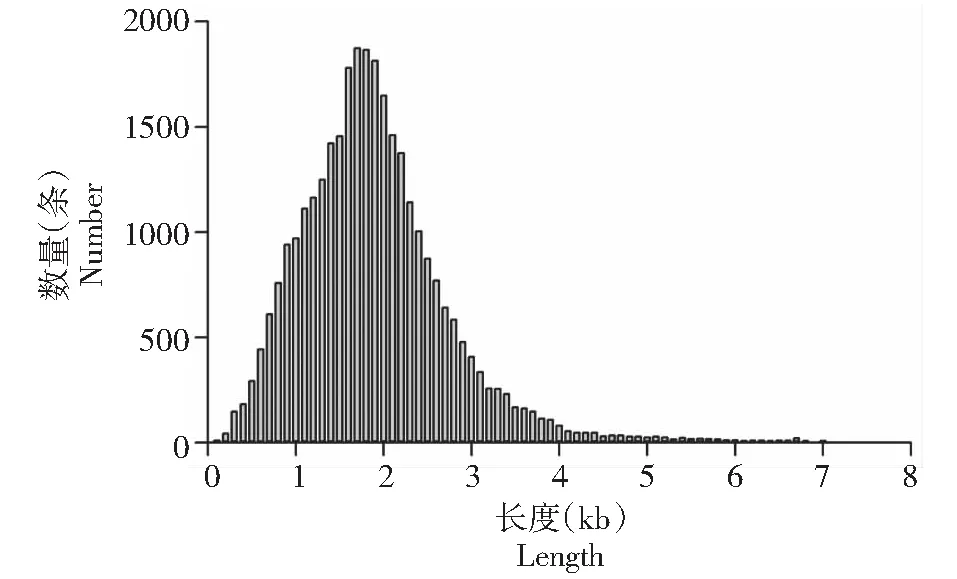
图1 红肉火龙果成熟茎的全长转录组获得的转录本长度分布
2.2 CDS预测
从表2可知,对30 313条高质量转录本进行CDS预测,获得CDS数量为40 255条。其长度最短为297 bp,最长为5202 bp,平均长度为926 bp,N50长度为1179 bp。统计其长度分布表明,CDS长度在200~600 bp的数量为14 372条,占35.70%;长度在800~1200 bp的数量为15 253条,占37.89%;长度为1200~2000 bp的数量有8456条,占21.01%;长度大于2000 bp 的数量为2172条,占5.40%。
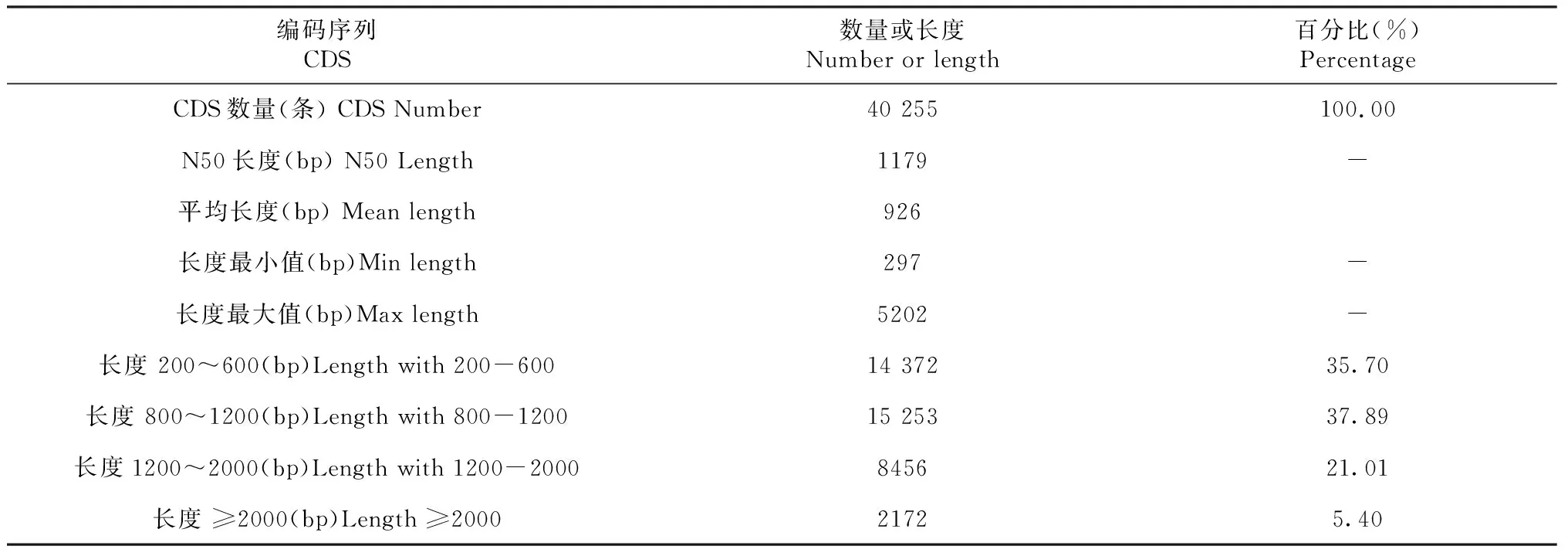
表2 预测CDS的数量或长度分布
2.3 序列注释
从表3看出,将30 313条高质量转录本在公共数据库中进行序列注释,共计有13 939条转录本被注释,占45.98%。在NCBI非冗余蛋白数据库(NR)中注释的转录本数量为13 789条,占所有转录本的45.49%。在NCBI核酸数据库(NT)中注释的转录本数量为10 777条,占所有转录本的35.55%。在Pfam数据库中注释的转录本数量为13 136条,占所有转录本的43.33%。在KEGG数据库注释的转录本数量为7582条,占所有转录本的25.01%。
从表4可知,统计注释到的相关物种上的转录本数量依次为甜菜(Betavulgaris)、菠菜(Spinaciaoleracea)、葡萄(Vitisvinifera)、克里曼丁橘(Citrusclementina)、麻疯树(Jatrophacurcas)和莲(Nelumbonucifera),其数量分别为9371条(50.95%)、4579条(24.90%)、436条(2.37%)、258条(1.40%)、205条(1.11%)和204条(1.11%)。
在GO数据库中共有13 059条转录本被注释到生物学过程、细胞成分和分子功能等三类,占所有转录本的43.08%(表3)。生物学过程中注释到24个类别,其中含有转录本最多的三类依次为细胞的过程(66.41%)、代谢过程(60.94%)和单一的生物过程(39.06%)。细胞成分中注释到22个类别,其中含有转录本最多的三类依次为细胞组分(82.03%)、细胞器(54.69%)和细胞器组分(35.94%)。分子功能中注释到19个类别,其中含有转录本最多的三类依次为结合(64.06%)、催化活性(51.56%)和核酸结合转录因子活性(6.25%)。
在KOG数据库注释的转录本数量为10 018条,占所有转录本的33.05%(表3)。从图2可知,注释为仅通用功能预测的转录本最多,数量为1701条,占16.98%;其次为翻译后修饰、蛋白质周转和伴侣蛋白,为1203条,占12.01%;其他较多的分别为信号转导机制(901条,8.99%),翻译、核糖体结构和生物发生(656条,6.55%),功能未知(580条,5.79%),碳水化合物的运输和新陈代谢(543条,5.42%),转录(530条,5.29%),RNA加工和修饰(526条,5.25%),细胞内运输、分泌和囊泡运输(518条,5.17%)。
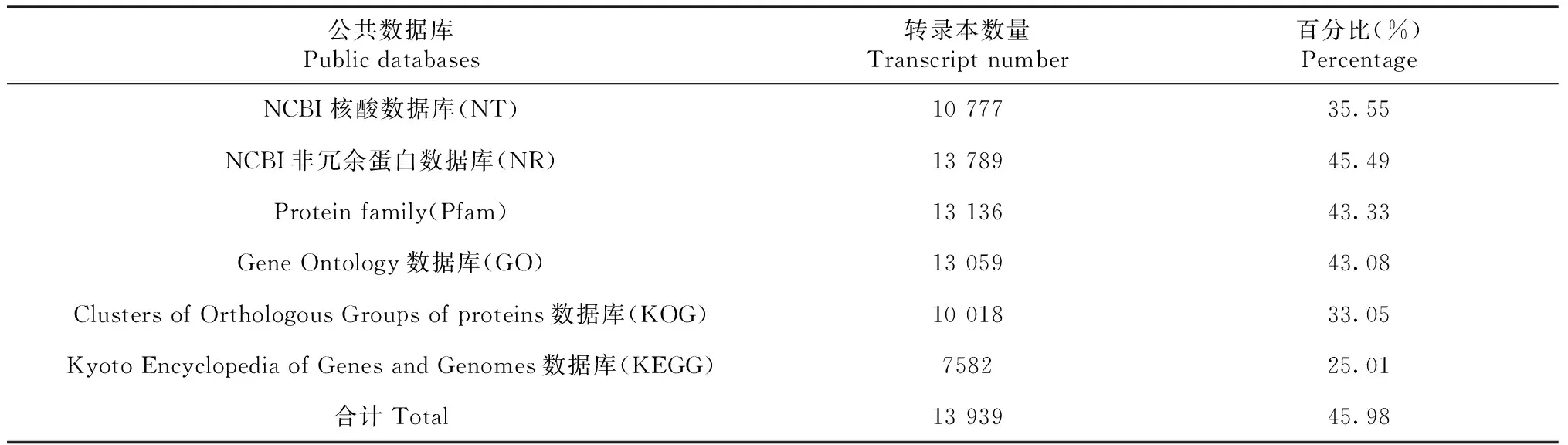
表3 红肉火龙果成熟茎全长转录组测序获得的转录本注释
2.4 转录因子家族预测
与植物转录因子数据库(Plant Transcription Factor Database,PlantTFDB)比对,共计有11 628条转录本注释为转录因子。统计转录因子家族表明,共含有56个家族,其含有转录本数量较多的家族如表5所示。MYB_related家族的转录本最多,数量为1162条,占10%;其次为bHLH家族(basic/helix-loop-helix),转录本数量为1066条,占9.17%。NAC(NAM、ATAF和CUC)家族、WRKY家族和FAR1家族含有的转录本数量依次为788、687和669条,分别占所有转录因子数量的6.78%、5.91%和5.75%。
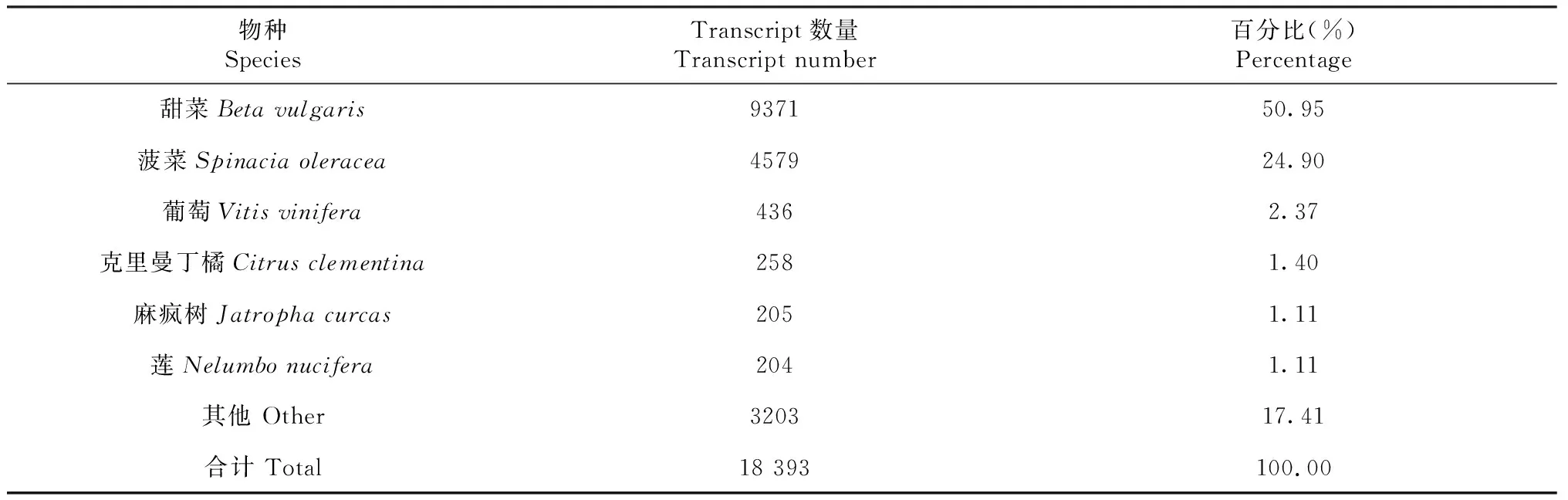
表4 NR数据库注释的物种数量

A:RNA加工和修饰;B:染色质结构和动力学;C:能源生产和转换;D:细胞周期控制、细胞分裂和染色体分割;E:氨基酸转运和代谢;F:核苷酸转运和代谢;G:碳水化合物的运输和新陈代谢;H:辅酶转运和代谢;I:脂质运输和新陈代谢;J:翻译、核糖体结构和生物发生;K:转录;L:复制、重组和修复;M;细胞壁/膜/包膜生物发生;N:细胞运动;O:翻译后修饰、蛋白质周转和伴侣蛋白;P:无机离子转运和代谢;Q:次生代谢产物的生物合成、转运和分解代谢;R:仅通用功能预测;S:功能未知;T:信号转导机制;U:细胞内运输、分泌和囊泡运输;V:防御机制;W:细胞外结构;Y:核结构;Z:碳骨架A: RNA processing and modification; B: Chromatin structure and dynamics; C: Energy production and conversion; D: Cell cycle control, cell division, chromosome partitioning; E: Amino acid transport and metabolism; F: Nucleotide transport and metabolism; G: Carbohydrate transport and metabolism; H: Coenzyme transport and metabolism; I: Lipid transport and metabolism; J: Translation, ribosomal structure and biogenesis; K: Transcription; L: Replication, recombination and repair; M: Cell wall/membrane/envelope biogenesis; N: Cell motility; O: Posttranslational modification, protein turnover, chaperones; P: Inorganic ion transport and metabolism; Q: Secondary metabolites biosynthesis, transport and catabolism; R: General function prediction only; S: Function unknown; T: Signal transduction mechanisms; U: Intracellular trafficking, secretion, and vesicular transport; V: Defense mechanisms; W: Extracellular structures; Y: Nuclear structure; Z: Cytoskeleton图2 转录本的KOG注释 Fig.2 KOG annotations of transcripts
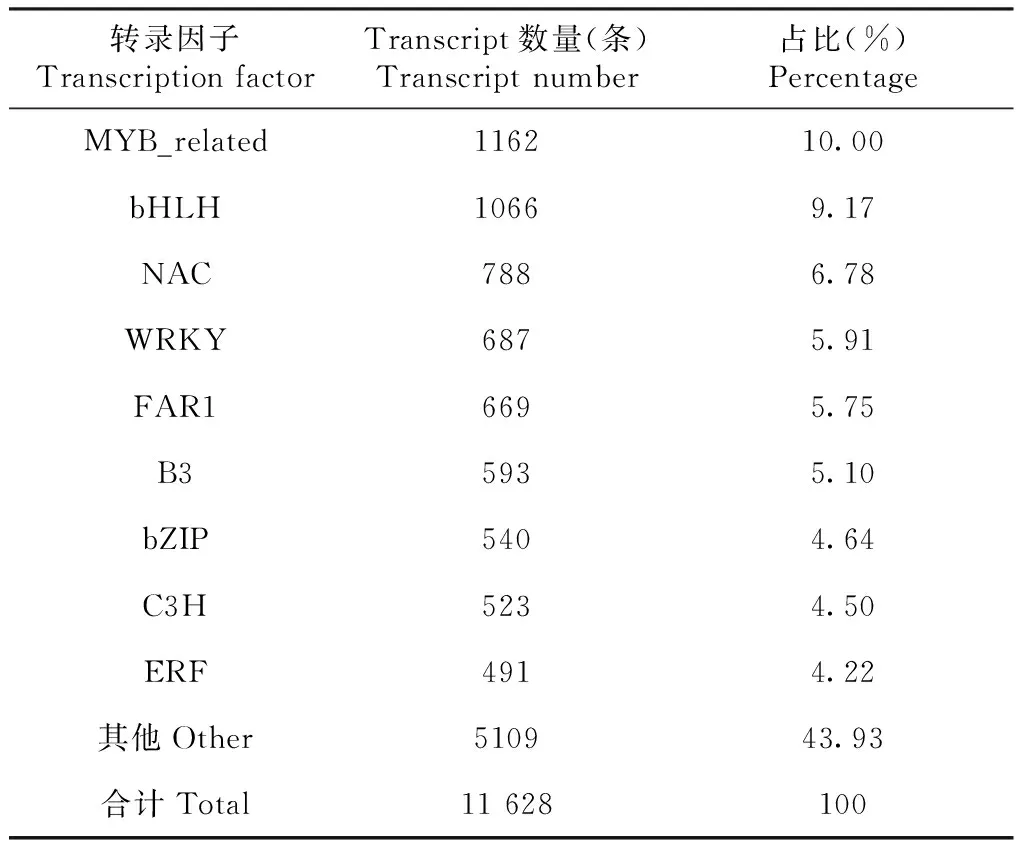
表5 预测的转录因子家族和数量
2.5 蔗糖代谢相关基因克隆
根据红肉火龙果成熟茎的全长转录组测序注释结果,筛选蔗糖代谢相关的基因(表6)。获得一条与菠菜蔗糖磷酸合酶(sucrose-phosphate synthase,SPS)具有81.03%相似性的转录本Transcript 920,其长度为3637 bp,含有3183 bp的CDS,编码2060个氨基酸。获得一条与红苋菜蔗糖合酶(sucrose synthase,SUS)具有93.90%相似性的转录本Transcript 3494,其长度为2719 bp,含有2 412 bp的CDS,编码803个氨基酸。获得一条与藜麦液泡酸性转化酶(acid beta-fructofuranosidase,AINV)具有72.61%相似性的转录本Transcript 7574,其长度为2229 bp,含有1935 bp的CDS,编码644个氨基酸。

表6 全长转录组测序获得的蔗糖代谢相关基因及序列相似性
此前针对红肉火龙果成熟茎和成熟果实果肉,利用基于Illumina平台的转录组测序进行分析,获得若干在茎中高表达的蔗糖代谢相关Unigene[11]。SPS相关的c49878_g1长度为464 bp,与Transcript 920序列相似性为100.00%,其在成熟茎中的表达量约为成熟果实的3倍(表7)。SUS相关的c21997_g1长度为614 bp,与Transcript 3494序列相似性为99.16%,其在成熟茎中的表达量约为成熟果实的45倍。AINV相关的c34087_g1和c24912_g1,长度分别为665和320 bp。与Transcript 7574序列相似性分别为99.06%和99.08%,在成熟茎中的表达量分别约为成熟果实的27和72倍。
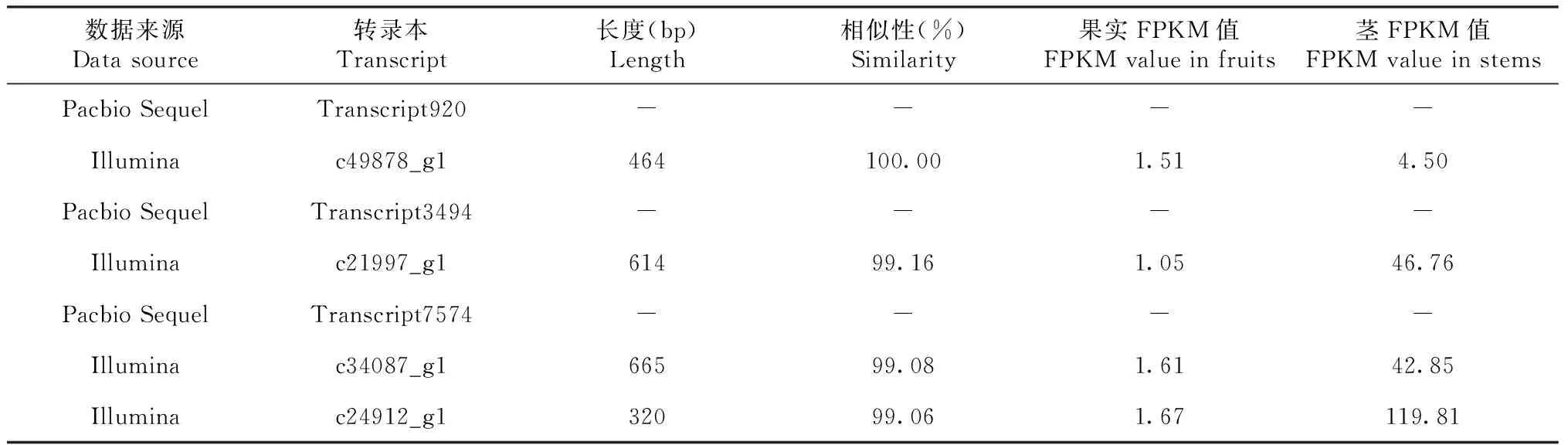
表7 蔗糖代谢基因与此前Illumina测序结果比较
3 讨 论
对缺乏基因组序列的非模式作物开展功能基因组学研究,其前提是获得基因全长序列。转录组测序能够快速获得大量基因的转录本序列,已在非模式作物中广泛应用。此前普遍利用基于Illumina平台的第二代高通量测序,其读长较短,极难获得基因全长序列。基于PacBio Sequel平台的第三代高通量测序具有显著的长读长优势,可用于全长转录组测序[14-15]。第三代高通量测序的主要缺点是错误率较高,表现为插入、缺失或错配,均为随机错误,可进行校正[16]。通过Subread聚类的自我校正,获得的CCS可作为转录本的参考序列[17]。
此前利用基于Illumina平台的转录组测序对红肉火龙果成熟茎和成熟果实果肉进行分析,获得Unigene平均长度为690 bp,长度大于1000 bp的Unigene仅占18.66%[11]。本研究获得30 313条高质量转录本,平均长度为1829 bp,显著长于Illumina测序。同时,长度大于1000 bp的转录本占比为88.96%,大于2000 bp占比为41.50%。由此可见,全长转录组测序获得的转录本长度明显长于Illumina测序,有利于获得基因的全长序列。
获得红肉火龙果成熟茎的30 313条高质量转录本,仅有45.98%的序列在常见公共数据库中被注释,注释到的物种主要为藜科(Chenopodiaceae)的甜菜和菠菜。与近期一些非模式作物的全长转录组测序相比,其转录本的注释率明显偏低。例如,对禾本科(Gramineae)薏苡属(Coix)薏苡(C.lacryma-jobiLinn.)苗期叶片开展全长转录组测序,转录本平均长度为2318 bp,约91.50%转录本被注释,物种为高粱、玉米、谷子、水稻和甘蔗等禾本科作物[18]。对蔷薇科(Rosaceae)栒子属(Cotoneaster)作物山东栒子(C.schantungensis)叶片、花和成熟果实进行全长转录组测序,约98.86%转录本被注释,物种为苹果、白梨、桃、梅、野草莓和沙梨等蔷薇科作物[19]。对桑科(Moraceae)木波罗属(Artocarpus)作物波罗蜜(A.heterophyllus)茎和叶进行全长转录组测序,获得转录本平均长度为1684 bp,约97.56%转录本被注释,物种为同属于桑科的川桑(Morusnotabilis)[20]。上述作物在同一科内均有其他作物的基因组序列发布,在其亲缘关系较近的情况下注释成功率极高。红肉火龙果属石竹目(Caryophyllales)仙人掌科,该科尚未有物种的基因组序列发布。目前仅石竹目藜科的甜菜和菠菜基因组序列发布,因而相关转录本的注释率较低。在未来仙人掌科作物基因组序列发布后,有必要对红肉火龙果成熟茎的转录本序列重新注释。
火龙果等仙人掌科作物的叶片退化为刺,其肉质茎是主要的光合器官。研究成熟茎的转录组模式,分离光合作用代谢途径的相关基因全长,有助于研究蔗糖的合成、贮藏和转运,进而调控果实品质形成。此前利用Illumina平台的转录组测序对红肉火龙果成熟茎和成熟果实果肉进行分析,仅获得若干参与成熟茎中蔗糖代谢和转运相关基因部分序列[11]。本研究获得蔗糖磷酸合酶SPS、蔗糖合酶SUS、液泡酸性转化酶AINV等均参与蔗糖合成和降解等代谢[21]。与Illumina转录组测序结果综合分析,其较短的Unigene序列与其相应的全长转录本具有99%以上的核苷酸序列相似性,说明来源于同一基因。这些较短Unigene的数字表达模式表明其主要在成熟茎中表达,推测其参与源器官中蔗糖代谢。下一步可根据全长转录组测序获得的基因全长克隆该基因,开展后续的功能验证等分析。
4 结 论
研究利用基于Pacbio Sequel平台的转录组测序对红肉火龙果成熟茎进行全长转录组测序分析,获得30 313个高质量转录本,其长度显著长于Illumina测序结果。全长转录组测序获得的高质量转录本,能结合Illumina测序获得的转录本序列和基因数字表达结果,快速筛选若干候选基因的全长序列,为开展基因功能研究提供有力基础。
