膝骨性关节炎合并骨质疏松症调节机制的研究进展
2022-04-19陈欣刘建军董万涛张兵刚李宁席芳琴宁学乾巩彦龙
陈欣 刘建军 董万涛 张兵刚 李宁 席芳琴 宁学乾 巩彦龙
1.甘肃中医药大学,甘肃 兰州 730000 2.甘肃中医药大学附属医院,甘肃 兰州 730000
膝骨性关节炎(knee osteoarthritis,KOA)和骨质疏松症(osteoporosis,OP)是与年龄增长相关的骨与关节退行性疾病[1]。在KOA的疾病进程中,膝关节发生的病理变化除了滑膜炎症、关节面软骨退变、半月板退变和韧带、肌肉的改变外,还包括软骨下骨改变和关节周围骨赘形成,即骨质和骨代谢改变也参与了KOA的病程[2]。KOA与OP的关系,目前仍存在较大的争议,但多数研究支持KOA与OP之间存在密切的相关性[3],两者在流行病、病理和治疗等方面均有着密切的关联,OP是KOA的危险因素,也能影响KOA的进展,KOA可加重OP[4]。研究[5]显示,KOA合并OP在我国的发病率高达30 %。目前,对于KOA的生物力学型、骨质疏松型、代谢型以及炎症型等表型的研究正逐渐展开[6],其中骨质疏松型已被多学科研究者广泛关注。KOA与OP两者通过复杂的机制调节相互影响、相互作用,本研究从以下几个方面展开分析,以期为膝骨性关节炎合并骨质疏松症调节机制和临床治疗提供理论基础。
1 炎症在KOA并OP中的调节作用
KOA虽属于非炎症性膝关节疾病,但是免疫系统多种成分都参与到KOA炎症活化的病变过程中[7]。因增龄、激素改变等诱发的骨代谢异常病程中,导致机体发生低度氧化应激而引起免疫系统的低度活化,免疫系统的低度活化导致机体处于系统性微炎症状态进而可诱发OP[8]。
KOA和OP并非免疫性疾病,但在两者病程中炎性因子参与了骨吸收和软骨降解。研究发现,核转录因子NF-κB(nuclear transcription factor-κB,NF-κB)因能刺激各种促炎细胞因子的产生(如TNF-α、IL-6等)而与OP和OA的发病及疾病进展相关[9],在KOA病程,促炎因子TNF-α能通过NF-κB信号促进炎症介质向膝关节滑膜、软骨等组织释放,引发膝关节软骨退变、破坏及降解基质,部分炎性因子(如IL-6)能加强炎性因子间的协同作用,对滑膜炎症的发生发展起到促进作用,诱导基质金属蛋白酶(matrix metalloproteinases,MMPs)和聚蛋白多糖酶-4/5(Aggrecanase-4/5,ADAMts4/5)的释放而降解软骨[10],促进软骨细胞外基质(extracellular matrix,ECM)降解,导致膝关节软骨结构破坏;同时TNF-α与IL-6还可通过诱导NF-κB受体活化子配体(receptor activator of NF-κB ligand,RANKL)的表达以促进骨髓破骨细胞的生成,从而抑制骨形成并增强骨吸收[11],在卵巢切除(OVX)小鼠模型中抑制NF-κB可预防骨质疏松性骨量丢失[12],给予OP大鼠补充乳铁蛋白后能抑制TNF-α与IL-6引起的骨量丢失[13]。Nur Adeelah等[14]在膝骨性关节炎-骨质疏松模型大鼠中发现NF-κβ、NF-α、IL-6表达均升高,可见上述证据都支持KOA与OP呈正相关。
炎症在KOA合并OP病程中的参与作用和调节机制较复杂,涉及到机体免疫机制、炎性细胞因子等,这些因素之间相互作用、相互影响,各作用平衡可维持正常的关节软骨和软骨下骨代谢,若失衡则将引起关节软骨和软骨下骨代谢异常。
2 生物力学在KOA并OP调节中的作用
适当的应力对膝关节软骨内环境的稳定、力学适应性、软骨下骨微结构稳定均有益处。机械过负荷导致的KOA是KOA表型中的一种亚型也称生物力学型KOA。生物力学失衡是引起KOA软骨病理变化的最初因素[15],其在KOA进展中起到了瀑布效应[16]。膝关节应力失衡导致软骨下骨的转换和吸收异常,在KOA早期软骨下骨的病变先于软骨退变,机体在自我调节、适应这种应力改变以稳定膝关节、保护关节软骨的同时继发性地引起软骨受力异常,因此也损坏了软骨;此外,应力失衡引起关节腔压强变化可引起软骨下骨吸收造成局部发生骨质疏松[17]。“膝关节不均匀沉降”理论认为,KOA患者因膝关节内外侧所受的应力不同,使胫骨平台内侧易发生骨质疏松性微骨折而出现沉降现象,为生物力学失衡在KOA合并OP中的作用提供了理论基础[18-19]。
生物力学在KOA与OP的发病及病程进展中起到了主要的作用,一方面,KOA与OP都是与年龄增长相关的骨与关节退行性疾病,增龄引起膝关节软骨下骨发生骨质疏松,这种软骨下骨质疏松因受膝关节生物力学、应力等的影响是非均匀的[20],进而导致关节软骨受力不均,出现不同程度的软骨下骨的骨小梁破坏、微循环损伤,引起关节面塌陷,骨质及膝关节稳定性等发生不可逆性损害,应力失衡后引起关节软骨的磨损、破坏,又可进一步累及软骨下骨[21-22],导致KOA的发生;另一方面,膝关节关节腔及其内外的化学、物理平衡破坏后,膝关节力学失稳、应力失衡而导致KOA的发生,同时随着年龄、免疫、内分泌等原因引起的性激素分泌不足、免疫系统活化,导致钙、磷、维生素D等物质的吸收与转化能力降低,这些因素导致了软骨下骨OP的发生,使得骨骼在骨的组织结构、骨的静力、动力等生物力学上的性能下降[23]。Liu等[24]观察了OP的骨组织病理微结构,结果发现除骨皮质变薄外,骨面及软骨面也在形态学和完整性方面发生了局部缺损、粗糙等变化,可见骨性关节炎往往与骨质疏松合并发病。
3 激素与骨代谢在KOA并OP中的调节作用
激素能够直接或间接的调节骨与软骨代谢,在KOA合并OP的进展中,甲状旁腺素(parathyroid hormone,PTH)、雌激素(estrogen)、降钙素(calcitonin,CT)等激素在抑制软骨退变、调节骨代谢(主要是骨的合成和吸收平衡)等方面具有重要作用,可以改善膝关节炎症环境、防止骨量减少而延缓疾病的进展[25]。
3.1 甲状旁腺素
PTH对KOA的作用,一方面是抑制关节软骨细胞的终末分化、防止软骨损伤而延缓KOA的病程[26],另一方面,其抑制滑膜细胞分泌IL-6等促炎因子,以减轻滑膜炎症引起的软骨基质降解,从而延缓或保护了软骨退变[27]。PTH的主要靶器官是骨、肾和小肠,其能促进破骨细胞(osteoclast,OC)活性加速骨吸收,也能促进1,25(OH)2D3的合成以加强肠钙吸收、促进骨对钙的重吸收,以防止骨量减少[28],小剂量或间断性升高的PTH能促进骨形成,而持续高浓度的PTH能促进骨吸收[29],研究发现由原发性甲状旁腺功能亢进症、甲状旁腺肿瘤等原因引起的PTH分泌过多会造成骨吸收而可能发生OP[30];PTH也是调节关节透明软骨生长的重要因子之一[31],在KOA的软骨病理变化过程中PTH可通过调节PTH/PTHrP1型受体(type1 PTH/PTHrP receptor,PTH1R)、激活丝裂原活化蛋白激酶(mitogenactivated protein kinase,MAPK)等多条途径调节软骨细胞分化,抑制软骨细胞肥大分化而凋亡,而调节软骨和骨代谢[32]。在KOA合并OP病程中PTH可防止软骨退变和软骨下骨退化,诱导软骨再生或软骨细胞增殖,有利于局灶性骨与软骨缺损进行修复;还可作用于软骨下骨前列腺素E2(prostaglandin E2,PGE2)以改善软骨下骨质量,保持膝关节稳定以减轻膝关节软骨损伤[33]。
3.2 雌激素
妇女绝经后随着雌激素(estrogen,ER)的减少,KOA等骨性关节炎和OP的发病率明显升高,说明雌激素对骨关节的作用是明显的[34],雌激素受体(estrogen receptors,ERs)也广泛存在于关节组织中[35],研究发现,芳香化酶基因和雌激素受体基因参与调节雌激素的分泌,两种基因的突变与下肢肥大性骨关节炎的严重程度相关[36]。雌激素及相关药物对绝经后早期KOA或OP性骨性关节炎患者具有治疗作用,可直接作用软骨下骨,也可直接和/或间接作用于关节软骨、滑膜和肌肉等关节周围组织。此外,研究发现雌激素、选择性雌激素受体调节剂(selective estrogen receptor modulators,SERMs)能降低骨转换[37],以防治软骨下骨微结构的破坏、维持骨密度及力学性能,进而增强了软骨下骨的骨强度,避免因软骨下骨的改变引起的对膝关节软骨和软骨下骨结构的损伤与破坏,从而减少骨质疏松性骨关节炎的发生。
3.3 降钙素
降钙素能抑制破骨细胞的分化和活性,以减少钙盐从骨释放、促进骨盐在骨的沉积,其还可减轻关节炎症而保护关节软骨[38]。在KOA合并OP中降钙素的调节作用主要表现在以下几方面,第一,保护软骨下骨,KOA早期其可抑制软骨下骨中破骨细胞活性,减少骨吸收而保持软骨下骨的强度,以防止因软骨下骨塌陷、硬化造成的膝关节不稳定或不平衡而导致的关节软骨的磨损、破坏,进而延缓了KOA的病程[39];第二,保护膝关节软骨,其与受体结合后作用于软骨细胞而在关节软骨中分泌足量的蛋白多糖和Ⅱ型胶原以保护关节软骨,其还可促进软骨下骨组织分泌足量的蛋白多糖以修复受损的关节软骨,起到保护关节软骨的作用[40];第三,抑制关节炎症,其能抑制膝关节滑膜等组织中白细胞介素1β(IL-1β)、基质金属蛋白酶(matrix metalloproteinase,MMPs)的合成与分泌,以减轻膝关节炎症反应;其能促进膝关节软骨细胞中转化生长因子-β(transforming growth factor-β,TGF-β)的合成[41],减少软骨细胞凋亡、吸收和软骨退变[42]。
参与调节KOA合并OP病理变化的还有糖皮质激素、生长激素、甲状腺素、瘦素等[43-44],这些激素之间及激素与机体之间相互作用、相互协调共同作用于膝关节软骨、调节骨代谢和骨转换。
4 遗传对KOA并OP的作用
遗传机制为生物体生命过程提供了表观遗传学修饰等在内的各种蓝图[45];表观遗传机制参与机体对环境的适应和对复杂疾病发病的调控[46],研究[47]显示,表观遗传机制(DNA甲基化、组蛋白尾部翻译后修饰、非编码RNA等)参与了软骨、骨细胞分化和机械转导,在KOA、OP等骨与关节疾病患者中已发现了各种表观遗传异常,但是它们的实际致病作用仍然不清楚[48]。在KOA合并OP过程中表观遗传学强调遗传和环境因素之间复杂的相互作用是该病的决定因素,此外他们还影响疾病的预后[49]。
Matlock等[50]应用DNA甲基化分析了KOA患者关节软骨和软骨下骨,结果发现了多个差异甲基化的胞嘧啶-磷酸-鸟嘌呤位点(cytosine phosphate guanine site,CpG)、诸多的差异基因在关节软骨及软骨下骨的相同区域共表达,在鉴定的7 361个CPG中有转化生长因子β-3(TGFβ-3)、活化T-细胞核因子1(NFATC1)、组蛋白去乙酰化酶(HDAC4)、同源盒基因A7 (HOXA7)等约2 800个基因在软骨和软骨下骨中同时存在。G蛋白偶联受体(G protein-coupled receptors,GPCRs)参与骨与软骨发育和重塑、炎症和免疫反应等多种生物学过程,是最丰富的跨膜蛋白家族,其中钙敏感受体基因(CaSR)、γ-氨基丁酸B型受体1(Gababr1)、G蛋白耦联受体C家族6组A(Gprc6A)、Tas1r3、代谢型型谷氨酸受体1(Grm1)等部分基因及其信号通路作用软骨与骨稳态,是调节骨关节发育和重建的必要物质,这些GPCRs的破坏或突变会导致KOA合并OP等人类骨骼疾病的发生、发展[51-52]。陈桐莹等[53]利用生物信息学方法分析了OP与KOA的关系,结果显示有miR-3202、miR-4687-3p、miR-4508、miR-320b四个差异的miR在OP与KOA中有交集,筛选出了参与调控OP与KOA的十个核心基因,即VEGFA、ESR1、CCND1、PRKACA、GSK3β、H2AFX、SHH、EGR1、DNMT3A、DNMT3B。
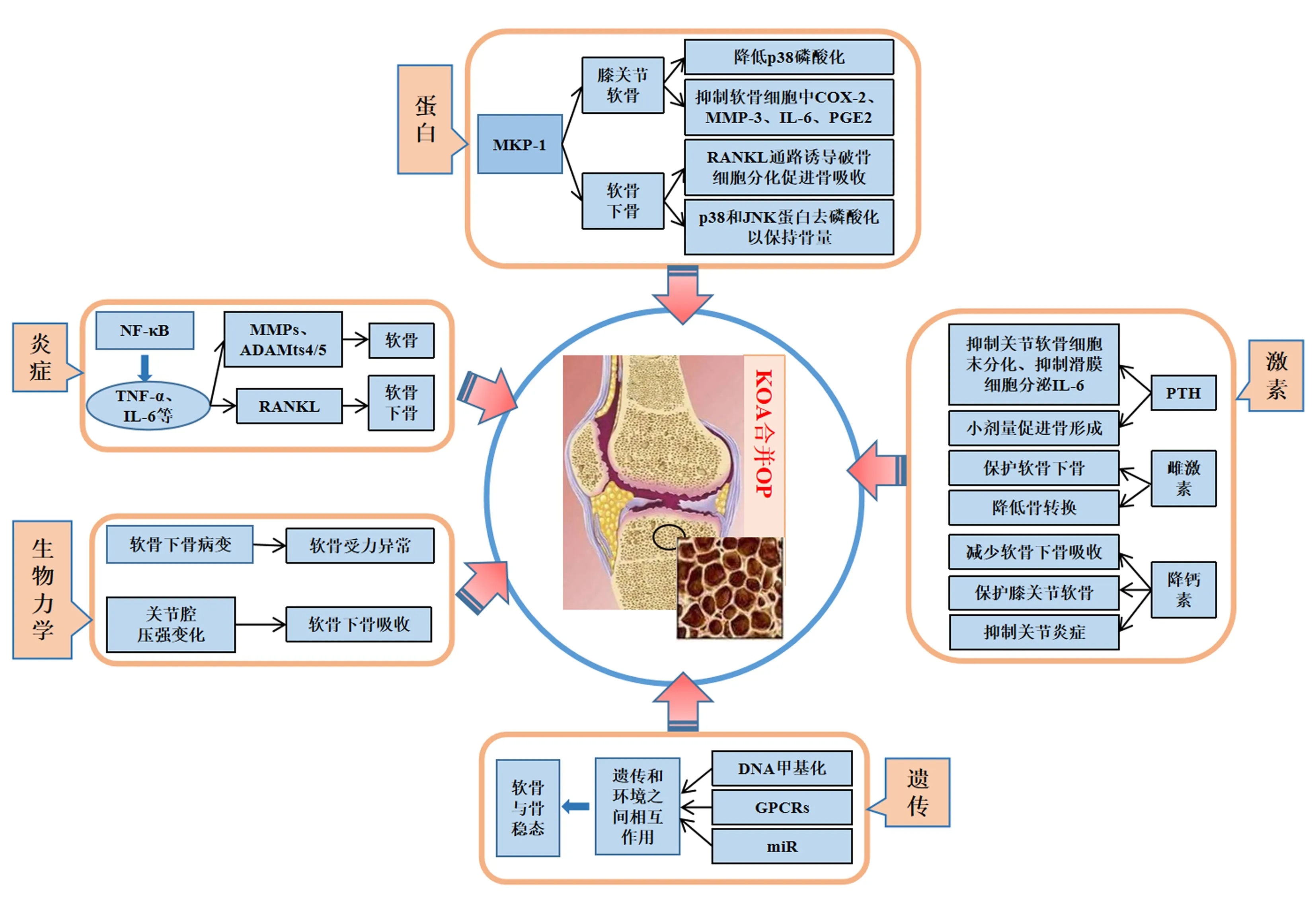
图1 KOA合并OP的调节机制示意图Fig.1 Schematic diagram of the regulatory mechanism of KOA incorporation of OP
此外,长正五聚蛋白PTX3(the long pentraxin PTX3)[54]、雌激素受体α(ERα)基因的多态性[55]等遗传及表观遗传机制在KOA合并OP的发病、病理过程中发挥了重要的调节作用。
5 蛋白在KOA并OP中的调节作用
蛋白质是构成生命、参与生命活动的重要物质,在组成关节的结构骨、软骨、韧带、神经、血管、肌肉中都存在多种功能各异的蛋白质,它们参与骨关节功能运动、提供能量、物质代谢、细胞增殖与分化、应激反应、炎症、凋亡和免疫防御,各种功能间相互协调共同维持关节的功能。
KOA与OP在临床中常存在共病的现象,其具有复杂多样的调控机制。时刚等[56]制备了新西兰大白兔KOA并OP模型,运用蛋白组iTRAQ技术定量定性分析了各组膝关节软骨蛋白组学,筛选出了KOA与OP共病组中差异明显,且与骨骼、肌肉、周围神经等组织系统和炎症、凝血、免疫等功能系统相关的8个蛋白,即α-2-HS糖蛋白、M型肌酸激酶、纤维蛋白原(β、γ链)、血清白蛋白、维生素D结合蛋白、asporin蛋白、髓鞘PO蛋白,这些蛋白可能是KOA并OP的基础物质。丝裂原活化蛋白激酶(mitogen2activated protein kinases,MAPKs)参与调节成骨、软骨代谢、脂肪代谢、能量转化等机体多种生物学过程,MAPK磷酸酶(MKPs)是MAPK信号传导的负调节因子,并使细胞外信号调节激酶(ERK)、p38和c-Jun N-末端激酶(JNK)三种MAPK去磷酸化,MKP-1是MKPs家族成员中对上述三种MAPK调节作用最明显的一个,其在KOA病程中作用主要是抑制关节炎症;MKP-1在软骨中的表达,降低了p38的磷酸化,同时抑制软骨细胞中环氧化酶-2(cyclooxygenase-2,COX-2)、基质金属蛋白-3(MMP-3)、IL-6和PGE2的产生[57];在KOA的治疗中其能协同非甾体抗炎药(NSAIDs)、类固醇药物、金制剂、透明质酸类药物的治疗作用,在抑制关节炎症、软骨退变等病理过程中发挥疗效[58];在OP病程中,MKP-1可通过RANKL信号通路诱导破骨细胞分化促进骨吸收,其能够使p38和JNK蛋白去磷酸化以保持骨量[59],MKP-1基因敲除后小鼠成骨细胞分化和矿化功能被抑制,导致小鼠骨量减少,说明表明MKP-1通过成骨细胞和破骨细胞的共同调节以维持骨量[60]。此外还有基质金属蛋白酶(MMP)、Sirtuins、FOxOs[61]、Sclerostin、β连环蛋白(β-catenin)、Dickkopf 1蛋白(DKK-1)、Ⅰ和Ⅱ型胶原羧基端交联肽(CTX-I/Ⅱ)[62]等多种蛋白通过多种途径参与了KOA并OP的调控。
6 小结与展望
KOA与OP在临床中常存在重叠、合并的情况,有着复杂的联系,并且OP会促进KOA的疾病进展、KOA也可能加剧OP的发生,两者在发病机制、病理变化、调节机制、临床治疗等诸多方面存在着共同点。KOA合并OP患者病程中涉及到炎症、生物力学、激素与骨代谢、遗传、蛋白等多种途径和作用的调节,这些因素之间相互发生作用,通过复杂的机制在KOA合并OP的进程中发挥重要的作用,以维持骨代谢平衡、减轻炎症、保护关节软骨、稳定膝关节,进而延缓膝关节的退变;针对不同的机制,已有对应的药物研发并在临床上使用,但是目前仍然没有可以阻止KOA并OP病情进展的药物。
KOA合并OP的早期,膝关节软骨下骨发生骨质疏松,引起软骨下骨微骨折,关节面塌陷导致膝关节生物力学失衡、应力发生改变而使软骨受力不均匀,加速了对关节软骨的磨损;引起软骨下骨微骨折引起局部循环破坏,使局部骨和软骨失去营养;激活了炎症、骨代谢、软骨代谢等多种机制,造成软骨损害、骨代谢失衡、继发性骨赘生成,最终引起膝关节功能障碍。但是KOA合并OP的调节机制仍有待进一步研究,随着研究的不断深入,阐明KOA合并OP病理过程中各种调节机制的生物学特性、生物学过程、变化规律、内在联系,将为KOA并OP防治提供新的思路和理论依据。
