脱屑性间质性肺炎一例并文献复习
2022-04-02蔡晓珊李颖于国华张绍坤岳贤文赵守香
蔡晓珊,李颖,于国华,张绍坤,岳贤文,赵守香
作者单位:261041山东省潍坊,潍坊市第二人民医院
脱屑性间质性肺炎(DIP)是特发性间质性肺炎的少见类型之一,其以肺泡腔内多量巨噬细胞聚集为主要特点,常被漏诊或误诊,临床-影像-组织病理综合分析以及病理医生对DIP病理特点有充足的认识对疾病的早期诊断和治疗具有重要意义。本文对近期确诊的1例73岁男性DIP患者进行报道,探讨其临床病理特征,以提高对该疾病的认识。
1 病例
患者男,73岁,因“胸闷、气短,间断发热半年”于2019年6月19日入院。患者半年前逐渐出现胸闷、气短,活动后加重,伴有发热,最高体温可达37.5℃,发热无明显规律。无胸痛咯血,无夜间阵发性呼吸困难及咳粉红色泡沫样痰。在外口服中药治疗,效果不佳,胸闷气短无减轻。患者自本次加重以来,饮食及睡眠可,大小便正常,体质量减轻6kg。既往体健,有吸烟史60年,每天20支,平素生活规律,无毒物、粉尘等接触史,其他既往史无特殊。体检双肺呼吸音低,双肺背部可闻及爆裂音,心率85次/min,心律规则,各瓣膜听诊区未闻及病理性杂音,腹平软,无压痛及反跳痛,双下肢无水肿。外院胸部CT考虑间质性肺炎,为求进一步明确诊断和治疗,来本院呼吸与危重症医学科住院。
入院后完善化验检查:二氧化碳分压34mmHg(1mmHg≈0.133kPa),白细胞计数9.82×109/L,血小板96×109/L,血沉60 mm/h,白蛋白34.5 g/L,降钙素原0.1 ng/ml,非小细胞癌胚抗原3.98ng/ml,痰液巨细胞病毒核酸检测阴性,痰液致病菌培养阴性,咽拭子涂片见少量革兰阳性球菌。2019年6月19日行肺功能检查,肺总量(FVC)为3.25 L,第1秒通气量(FEV1)为2.39 L,提示为小气道功能障碍,弥散量降低。入院后复查胸部CT(图1)示双肺胸膜下见近对称分布的磨玻璃影,病变以双肺上叶分布为著,部分区域内见多发薄壁囊腔影;左肺下叶外基底段见实性结节,边界清晰,边缘见浅分叶。2019年6月22日,患者在全身麻醉下行电子支气管镜检查,左、右侧支气管黏膜肥厚充血,各叶段支气管通畅,所视范围内未见明显异常。于右上叶支气管给予0.9%氯化钠注射液60ml灌洗,行灌洗液细胞学检查,支气管肺泡灌洗液(BALF)无色透明,细胞分类计数:细胞总数2.80×104/ml,其中肺巨噬细胞占10.75%,中性粒细胞占87.25%,淋巴细胞占1.75%,嗜酸性粒细胞占0.25%,中性粒细胞比例明显增高。灌洗液行鼻病毒、呼吸道合胞病毒核酸检测均为阴性,灌洗液查TB-DNA阴性,致病菌培养阴性。于右上叶后段支气管行支气管镜肺活检(TBLB),送病理检查。病理镜下见肺泡正常结构存在,肺泡间隔略增宽,间质内见少许淋巴细胞及中性粒细胞浸润,肺泡腔内见多量巨噬细胞聚集(图2~3)。

图1 治疗前轴位胸部CT肺窗表现:双肺胸膜下见近对称分布的磨玻璃影,病变以双肺上叶分布为著,部分区域内见多发薄壁囊腔影

图2 肺组织肺泡正常结构存在,间质少量淋巴细胞、中性粒细胞浸润(HE染色,×100)
经本院多学科诊疗团队(MDT)讨论,结合患者病史、影像学资料及BALF细胞分类计数和病理结果,认为患者间质性肺炎符合DIP。入院后给予多索茶碱平喘对症处理。明确诊断后,临床医师嘱其戒烟,未使用糖皮质激素等药物治疗,于2019年6月26日出院。出院继续戒烟,并于2019年7月29日复查,患者胸闷、气短现象明显改善,无发热。化验检查提示白细胞计数8.79×109/L,血小板168×109/L,血沉43 mm/h,白蛋白36.9 g/L,降钙素原0.068ng/ml。复查肺功能,FVC为3.68L,FEV1为2.64L。复查胸部CT双肺部分区域磨玻璃影明显吸收,囊腔影残留(图4),患者病情明显好转,后续继续随访中。
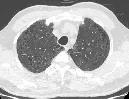
图4 治疗后轴位胸部CT肺窗表现:双肺磨玻璃影明显吸收好转,薄壁囊腔影残存
2 讨论
DIP是一种少见的特发性间质性肺炎,以肺泡腔内大量巨噬细胞聚集为主要特征性改变。DIP最早由Liebow等[1]在1965年提出,当初误认为肺泡腔内脱落聚集的是肺泡上皮细胞,并因此而得名。后经电镜研究证实,肺泡腔内聚集的细胞大多数为巨噬细胞及少许肺泡上皮细胞,但至今仍然沿用此名。由于DIP和呼吸性细支气管炎伴间质性肺病(RB-ILD)经常出现在吸烟者的肺组织中,被认为是主要影响吸烟人群的连续性疾病谱。2013年美国胸科学会和欧洲呼吸学会将RB-ILD和DIP归类为吸烟相关性特发性间质性肺炎(SR-IIP)。DIP为少见病,其发病率目前没有确切的流行病学统计数据[2]。其精确患病率可能高于目前文献的报道,因为该疾病临床症状及影像学改变均不具有特异性,容易造成误诊或漏诊,外科肺活检或经支气管冷冻活检是诊断的“金标准”[3]。DIP的主要病理特征是肺泡内存在大量巨噬细胞,这些巨噬细胞在整个肺泡内呈弥漫性分布[1],有丰富的嗜酸性胞浆,通常含有浅棕色的色素颗粒[4],也被称为“吸烟者的巨噬细胞”;有时也可以看到少许多核巨细胞[1]。虽然肺泡间质内有轻微的慢性炎症浸润,但肺泡结构通常保持良好,也可见到中等数量的嗜酸性粒细胞及淋巴细胞聚集,间质纤维化通常较轻[5]。对于晚期疾病,转化生长因子1的产生可导致肺泡间质纤维化。然而,与普通型间质性肺炎(UIP)不同,成纤维细胞灶几乎不会出现[3]。本例患者肺泡正常结构未被破坏,肺泡腔内见多量巨噬细胞聚集,间质可见散在的少许淋巴细胞及中性粒细胞浸润,病理学改变符合DIP。
DIP典型的发病年龄是40~60岁[5-6],也有儿童发病的报道[7]。男性多见,约是女性的2倍[8]。大多数为亚急性起病或隐匿,目前发病原因尚不明确,多数认为DIP是一种吸烟者的疾病,据报道与吸烟的相关性接近90%,但也与其他接触和疾病状况有关[3],包括:吸食大麻;暴露于铜、铍等;职业性接触灭火器粉末和柴油烟雾;纺织工人用尼龙长丝;某些药物的使用(西罗莫司、呋喃妥因等);自身免疫性疾病(硬皮病、类风湿性关节炎等);肺移植后DIP复发;某些传染病病原(如曲霉菌、丙型肝炎、巨细胞病毒)以及代谢性疾病(如戈彻病)等[3]。儿童发病原因常为其表面活性蛋白基因突变导致功能障碍[3,9]。常见症状是劳累时呼吸困难和干咳[5-6],部分有少量黏痰或痰中带血,约50%有胸痛或胸骨后疼痛,严重者体质量明显减轻,且有疲劳、肌痛、多汗等表现[10]。DIP的体格检查也缺乏特异性,约60%的患者出现吸气相爆裂音,近一半的患者会出现杵状指。肺功能检查显示明显的限制性通气功能障碍,弥散功能下降50%或以上,肺容积对称性减少[5,11]。DIP的HRCT改变也不特异,常表现为磨玻璃样结节,可为周围性、斑片状或弥漫性分布[12];周围胸膜下和基底部磨玻璃样影最常见[13],常分布均匀,无蜂窝状改变,这也是区别于UIP或特发性肺纤维化(IPF)的两个特征。在进行性疾病中,可以看到囊腔影和牵拉性支气管扩张,分布不完全在胸膜下[3]。纤维化不像非特异性间质性肺炎(NSIP)或UIP那样常见。DIP报道病例较少,因其临床表现和影像学特征均不具有特异性,确诊需要依靠临床-影像-病理多学科综合分析,最终取得组织标本,组织病理学镜下查见肺泡腔内多量巨噬细胞聚集为确诊依据。本例患者因胸闷、气短,活动后加重,伴有发热为初始症状就诊,无毒物、粉尘等接触史,有吸烟史,起病原因符合吸烟所引起;听诊双肺背部可闻及爆裂音,肺功能检查显示弥散量降低,比弥散正常;影像学改变为双肺上叶胸膜下近对称分布的磨玻璃影,内见多发囊腔影。结合临床表现-影像学资料-组织病理学特点,可以确诊为DIP。
鉴别诊断:(1)RB-ILD:RB-ILD与DIP均属于SR-IIP,在临床病史、影像学及组织学上都具有相似性。RB-ILD发病年龄平均为36岁,约2/3的患者高分辨CT扫描显示网状结节影或小叶中心磨玻璃结节[14],磨玻璃影常缺乏[8]。RBILD反映了吸入性暴露,其病变集中在伴有支气管周围纤维化和炎症的细支气管周围。而DIP影响气道并延伸至肺泡腔[14]。病理学特征方面,两种疾病均有大量含色素的巨噬细胞聚集,不同之处在于RB-ILD相对局限于呼吸性细支气管及其周围的气腔,远端肺泡腔通常不受累,且常有明显的呼吸性细支气管炎。(2)UIP:UIP和DIP都属于特发性间质性肺炎的范畴,影像学上UIP的重要特征是自上肺至下肺逐渐加重呈梯度胸膜下对称分布的蜂窝影,大部分病灶内可见扭曲增宽的支气管影,病变与正常肺组织分界清晰[8]。病变时相不一致,分布不均匀,病理学改变有成纤维细胞灶形成、间质的炎症、纤维化和蜂窝变组成的不同时相病变,而无肺泡腔内大量巨噬细胞的聚集。DIP病变常均匀分布,纤维化常不明显,无蜂窝肺改变[3],肺泡腔内有大量含色素的巨噬细胞聚集。(3)NSIP:双肺胸膜下对称分布的磨玻璃影,以双肺下叶分布为著,部分内见蜂窝征。病理学改变常为肺间质不同程度的炎性改变和纤维化,主要为小淋巴细胞的浸润,可见少量的浆细胞,病变呈片状或弥漫分布,时相一致[8]。局部肺泡腔内常含有多量泡沫细胞。(4)肺朗格汉斯细胞组织细胞增生症(PLCH):PLCH也是吸烟相关性肺疾病的一种,影像学表现早期常为大小不等的实性结节或囊腔结节,晚期往往是形态不规则的薄壁囊腔,甚至是蜂窝肺,病灶分布往往以中上肺叶分布为著,双肺下叶近膈顶区很少受累[15]。病理特点以朗格汉斯细胞组织细胞增生为主要特点,在高倍镜下有明显的核沟。(5)其他疾病(如UIP、NSIP、重度吸烟者,肺恶性肿瘤等)中,局部肺泡腔内亦有局灶性聚集的巨噬细胞,称为DIP样反应,不能误认为是DIP,因为这类巨噬细胞的聚集仅是局灶性的改变,而DIP肺泡腔内巨噬细胞聚集是弥漫均匀一致的。因此单纯靠临床、影像学或病理医生任何一方面来确诊DIP都有局限性和不准确性,多学科会诊具有重要的意义。尤其是对于小活检标本,病理医生做诊断前要结合临床及影像学改变来综合分析。
DIP的预后不一,10年总生存率为70%~88%[16]。DIP确诊后最重要的干预措施是戒烟。如果与特定职业暴露相关,避免暴露也是预防疾病进展的关键[17]。成人持续吸烟被认为是预后不良的指标[14,16]。戒烟后又复吸是导致疾病恶化的可能原因之一[10]。另外,儿童患者(尤其是伴有ABCA3突变的)预后较差[18]。大多数患者在使用皮质类固醇和免疫抑制疗法(硫唑嘌呤是最常用的药物)后,可保持病情稳定或改善,甚至完全康复[19]。也有文献报道约25%的患者在接受皮质类固醇治疗后,病情可能会继续进展,导致肺纤维化[20]。有严重进展性疾病的患者,可以进行肺移植[21],但是肺移植后仍有可能复发[22]。
综上所述,DIP目前的发病原因不明,约90%与吸烟相关;影像学往往表现为双肺胸膜下对称分布的磨玻璃影,部分内见薄壁囊腔;病理学典型改变为大量巨噬细胞在肺泡腔内弥漫性分布。临床-影像-病理的多学科综合分析对疾病的诊断意义重大。
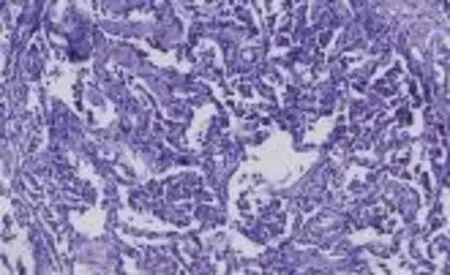
图3 肺泡腔内见多量巨噬细胞聚集,胞浆嗜酸性,内含浅棕色色素颗粒(HE染色,×200)
作者贡献声明:李颖与蔡晓珊对本文具有同等贡献;岳贤文与赵守香对本文具有同等贡献。
