线粒体质量控制失调与特发性肺纤维化的关系研究进展
2022-04-01冯同高瑕王波李万成
冯同,高瑕,王波,李万成*
1成都医学院,成都 610500;2成都医学院第一附属医院呼吸与危重症医学科,成都 610500
特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)是一种严重影响肺通气与换气功能的慢性下呼吸道疾病,表现为异常的肺间质纤维化及炎症,包括肺泡结构的破坏。IPF多发生于中年及老年人,平均诊断年龄为66岁,中位生存期为确诊后3~5年。IPF年发病率为(4.6~16.3)/10万,且呈上升趋势;患病率为(13~20)/10万,男性患者比例较高(男:女=1.5~1.7:1)。关于IPF的发病机制,目前被较多学者支持的是上皮-间充质转化(epithelial-tomesenchymal transition,EMT)。IPF被认为是肺泡上皮细胞功能异常及凋亡的结果,该损伤引起间充质细胞的迁移、增殖、活化,并形成了成纤维细胞及肌成纤维细胞病灶。激活的肌成纤维细胞分泌过多的细胞外基质,破坏了肺部结构[1]。目前,美国食品药品管理局(Food and Drug Administration,FDA)批准用于治疗IPF的药物有吡非尼酮及尼达尼布,但这两种药物的疗效有限,仅能适度缓解疾病的症状,不能改善IPF患者的生活质量及降低死亡率[2]。
线粒体除了具有能量供给的功能外,还在信号传导、新陈代谢及细胞死亡的调控中起重要作用[3]。线粒体质量控制通常通过三种不同的机制来维持:(1)线粒体生物合成;(2)线粒体动力学(融合与分裂);(3)线粒体自噬。广义的线粒体质量控制还包括线粒体内活性氧水平的调控,线粒体基质的蛋白酶、泛素蛋白酶体系统及线粒体衍生的囊泡。机体通过这些机制生成新的线粒体,限制损伤与衰老的线粒体过度积累,减少活性氧(reactive oxygen species,ROS)的产生,从而维持线粒体的动态平衡。本文探讨了包括线粒体ROS调控在内的线粒体质量控制失调与IPF的相关性,并针对线粒体质量控制提出IPF的防治策略。
1 线粒体质量控制
线粒体通过线粒体质量控制,即通过融合/分裂改变其形状及大小,通过线粒体自噬包裹受损的线粒体并将其传递至溶酶体进行清除,通过生物合成新生线粒体以补充线粒体池,通过线粒体内抗氧化系统降低ROS对细胞造成的氧化损伤,这些过程相互协调作用以适应不同条件下细胞新陈代谢的变化。
1.1 ROS水平的线粒体质量控制 ROS是一类含活性氧原子的反应性化学物质,包括超氧阴离子、羟基自由基、过氧化氢、臭氧及单线态氧等[4]。过多的ROS累积可造成线粒体的氧化破坏,同时,ROS作为信号分子参与了线粒体质量控制的调节,激活抗氧化系统。细胞内含有的抗氧化系统负责清除过多的ROS,维持氧化还原状态的稳定,阻止ROS诱导的氧化应激对线粒体DNA(mtDNA)及蛋白质的破坏。
1.2 线粒体动力学对线粒体质量控制的调控
线粒体分裂通过胞质动力相关蛋白1(dynamin relatedprotein 1,DRP1)、分裂蛋白1(fission protein 1,FIS1)、线粒体分裂因子(mitochondria fission factor,MFF)及线粒体动力蛋白49/51(mitochondrial dynamics proteins of 49 and 51,MID49/51)的作用完成。当位于细胞质或内质网中的DRP1被募集到线粒体外膜上时,DRP1与线粒体外膜上的受体FIS、MFF、MID49/51相互作用形成复合物,与DRP1结合的GTP的水解会收缩DRP1,使线粒体膜分裂,导致两个独立的子代线粒体形成,从而发生裂变[5]。线粒体外膜融合是通过线粒体融合蛋白(mitofusin 1/2,MFN1/2)及视神经萎缩相关蛋白1(optic atrophy 1,OPA1)进行的。OPA1在YME1样ATP酶(YME1-like 1 ATPase,YME1L1)的调控下,形成长式及短式的寡聚体,它们之间保持平衡,维持线粒体内膜的融合[6-7]。
1.3 线粒体自噬对线粒体质量控制的调控 线粒体自噬相关蛋白可分为E3泛素连接酶PARKIN依赖型及非PARKIN依赖型。其中最经典的途径为E3泛素连接酶PARKIN依赖型,需要磷酸酶及张力蛋白同源物诱导的蛋白激酶1(PTEN induced putative kinase 1,PINK1)的参与。正常情况下,PINK1位于线粒体内膜,由线粒体特异性蛋白酶线粒体内膜早老素相关菱形样蛋白(presenilin associated rhomboid like protein,PARL)及加工肽酶(mitochondrial processing peptidase,MPP)降解[8-9]。在线粒体损伤的情况下,PARL及MPP失活,抑制了PINK1由线粒体外膜转运至线粒体内膜的进程,导致PINK1在线粒体外膜上累积。位于线粒体外膜的PINK1的激酶结构域不仅可磷酸化线粒体外膜蛋白,还可募集及激活E3-泛素连接酶PARKIN[10]。激活的PARKIN将泛素化线粒体外膜及细胞质蛋白,泛素化蛋白与衔接子蛋白SQSTM1/p62及微管相关蛋白1轻链3(MAP1LC3/LC3)结合形成自噬体,启动自噬机制降解损伤严重的线粒体。
1.4 线粒体生物合成对线粒体质量控制的调控线粒体生物合成是一个高度协调的过程,利用线粒体及核编码基因来改变线粒体的大小及质量。它需要线粒体、细胞核、内质网及其他细胞器的协同作用。线粒体生物合成中关键的调控分子是过氧化物酶体增殖物激活受体-γ共激活因子-1α(PGC-1α),但其他转录因子,包括5'腺苷单磷酸激活蛋白激酶(5'-AMP activated protein kinase,AMPK)及核呼吸因子1/2(nuclear respiratory factor 1/2,NRF1/2)也参与其中[11]。PGC-1α作为共转录因子,转位至细胞核与NRF1/2结合,转录激活线粒体转录因子A(mitochondrial transcription factor A,TFAM)的表达,从而驱动mtDNA的转录及复制。
2 线粒体质量控制失调与IPF发病的相关性
目前,已有研究证据表明IPF与线粒体质量控制失调密切相关。人们已经认识到线粒体功能障碍是IPF及其他肺部疾病如慢性阻塞性肺疾病、哮喘发病机制的关键环节[12-14]。
2.1 线粒体氧化应激在IPF发病中的作用 ROS是参与细胞正常分化、增殖等过程的反应性物质,在病理状态下,线粒体内产生过多的ROS,不能被抗氧化系统及时清除,就会引发一系列反应,如mtDNA损伤、通透性转换孔开放及膜通透性改变,导致肺纤维化的发生[15]。研究发现,过量的线粒体ROS(mtROS)产生及蛋白酶复合体Ⅰ、蛋白酶复合体Ⅳ活性下降与纤维化小鼠模型中Ⅱ型肺泡上皮细胞(alveolar epithelial cells,AEC)的线粒体功能障碍有关[16-17]。与野生型小鼠相比,线粒体过氧化氢酶高表达转基因小鼠接触石棉、博莱霉素后肺纤维化的程度降低,小鼠Ⅱ型AEC中的mtROS水平也降低[17]。NRF2是一种氧化剂敏感的转录因子,可通过激活抗氧化系统,调节谷胱甘肽、硫氧还蛋白和还原型烟酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide phosphate,NADPH)的生物合成、利用及再生,并能控制线粒体及NADPH氧化酶的ROS生成,在维持细胞氧化还原稳态中起着至关重要的作用[18]。NRF2基因敲除小鼠肺部多种抗氧化酶表达降低,从而易发生ROS诱导的肺纤维化损伤[19]。同时,mtDNA损伤与氧化损伤及肺纤维化有关。IPF患者中mtDNA活性氧水平上升[20]。DNA碱基切除修复酶8-氧鸟嘌呤DNA糖基化酶1(8-oxoguanine DNA glycosylase 1,Ogg1)对修复Ⅱ型AEC氧化损伤起重要作用[21]。与野生型小鼠相比,Ogg1基因敲除小鼠石棉暴露后的肺纤维化程度、Ⅱ型AEC mtDNA损伤及氧化剂诱导的细胞凋亡增加[22]。线粒体哺乳动物的沉默信息调节因子3(silent information regulator 3,SIRT3)是烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide,NAD)依赖的sirtuin家族的成员,参与肺纤维化的调节。Sirt3基因敲除衰老小鼠会发生多个器官的组织纤维化,包括心脏、肝脏、肾脏及肺部,且IPF患者肺内SIRT3的活性下降[23]。SIRT3通过Ogg1及锰超氧化物歧化酶(Mn superoxide dismutase,MnSOD)调节蛋白脱乙酰化来减少mtDNA损伤,SIRT3缺乏可导致线粒体蛋白的乙酰化及失活,增强ROS引起的mtDNA损伤及细胞凋亡,促进肺纤维化[23]。因此,线粒体ROS增多是肺纤维化病理改变的机制之一。
2.2 线粒体动力学在IPF发病中的作用 线粒体是动态的细胞器,通过不断的融合与分裂,交换损伤的蛋白质及mtDNA,保持着动态平衡。小鼠Ⅱ型AEC中缺乏MFN1及MFN2会导致自发性肺纤维化的发生率及病死率增高。MFN1缺失导致线粒体破碎,MFN2缺失导致线粒体肿胀。在暴露于博来霉素的小鼠中,Ⅱ型AEC细胞的MFN1、MFN2及DRP1的表达水平增高,以减轻肺纤维化损伤[24]。G蛋白偶联受体C族5型A组(G protein-coupled receptor C group 5 type group A,GPRC5A)是G蛋白偶联受体家族的一个成员。在IPF损伤AEC中,GPRC5A的减少会导致线粒体融合蛋白MFN2及OPA1缺失,从而增加博来霉素所致肺纤维化的胶原蛋白沉积,刺激成纤维细胞的增殖与激活[25]。线粒体的融合及分裂参与了细胞凋亡的调节,线粒体融合可抑制细胞凋亡,而线粒体分裂可促进细胞凋亡。Ⅱ型AEC的凋亡是肺纤维化发生发展的关键因素。研究表明,miR-30a表达上调可减少Ⅱ型AEC的凋亡,抑制线粒体分裂,并降低DRP1的表达及转运,减轻肺纤维化的程度[26]。上述研究提示,线粒体动力学与肺纤维化之间存在密切关系,线粒体动力学改变可能通过影响线粒体活性氧生成、mtDNA分布、细胞内钙离子稳态及细胞凋亡等影响细胞能量供给及信号转导,从而促进肺纤维化的发生发展。
2.3 线粒体自噬在IPF发病中的作用 线粒体自噬是降解大分子蛋白质或细胞器的过程。当线粒体受损时,自噬体可将其包裹分隔,并递送至溶酶体进行清除及再回收[27]。PINK1缺乏会导致线粒体肿胀、功能障碍,并促进衰老相关的肺纤维化[12]。博莱霉素诱导的PARKIN基因敲除小鼠的线粒体自噬不足,无法清除受损线粒体,肺纤维化程度增加[28]。在IPF成纤维细胞灶的肌成纤维细胞和肺成纤维细胞中,PARKIN表达水平降低[29]。转录激活因子3(activating transcription factor 3,ATF3)是PINK1基因表达的负调控因子,其过表达可抑制PINK1启动子的活性,导致去极化线粒体的积累,线粒体ROS的产生增加。在ATF3缺失小鼠中,博来霉素诱导的肺纤维化程度减轻[30]。总之,目前动物实验及人体研究结果都表明促进自噬可以预防肺纤维化。
2.4 线粒体生物合成在IPF发病中的作用 线粒体生物合成是一个调控新生线粒体形成的生物学过程,主要涉及mtDNA的复制、mtDNA编码蛋白的合成以及协调线粒体动力学等作用。
线粒体生物合成能力下降与IPF的发病机制有关。在IPF患者与博莱霉素诱导的肺纤维化模型中,PGC-1α表达水平降低,此外,PGC-1α基因敲除小鼠经博莱霉素诱导后肺纤维化程度加重[31]。甲状腺激素(thyroid hormone,TH)可通过改善线粒体功能及形态、恢复线粒体膜电位来减轻博来霉素诱导的肺纤维化。IPF患者肺组织中碘甲腺原氨酸脱碘酶Ⅱ(recombinant deiodinase iodothyronine typeⅡ,DIO2,一种激活TH的酶)的活性及表达水平高于对照组,且与疾病严重程度相关。DIO2基因敲除小鼠经博来霉素诱导后肺纤维化程度加重。在博来霉素暴露后,TH促进了体内及体外AEC中的线粒体生物合成,改善了线粒体生物能,并减少了线粒体调节的细胞凋亡。在PGC-1α或PINK1基因敲除小鼠中,TH不能抑制肺纤维化,提示TH的作用是依赖于PGC-1α和PINK1而实现的[32]。AMPK是细胞生物能的关键传感器,可控制生物体内合成代谢的转换。在IPF患者及IPF小鼠模型中,AMPK活性在代谢活跃且凋亡抵抗的成纤维细胞相关的纤维化区域较低。AMPK激活物AICAR可上调TFAM并增加线粒体电子传递链复合物主要成分NADH泛醌脱氢酶1β亚复合物8的表达,增强线粒体的生物合成[33]。目前认为线粒体生物合成受损导致线粒体质量降低是肺纤维化线粒体功能障碍的另一重要内在机制。
3 以线粒体质量控制为靶点的IPF干预策略
恢复线粒体质量控制的干预措施可能是IPF的保护策略,如MitoQ作为线粒体靶向抗氧化剂,可减弱IPF肺成纤维细胞中TGF-β1及NOX4的表达[34]。研究表明,17β-雌二醇通过细胞核或线粒体雌激素受体对线粒体功能的调节可诱导抗氧化反应以及NRF1/2、PGC-1α的激活,从而促进线粒体的生物合成[35]。众所周知,IPF的AEC中PINK1缺乏是引发线粒体功能障碍的主要因素。因此,增强PINK1活性的药物可作为IPF的一种治疗选择。三磷酸激动素(kinetin triphosphate,KTP)可增强PINK1、PARKIN的活性并降低细胞凋亡标志物的表达,是抗肺纤维化的潜在药物[36]。同样,线粒体蛋白SIRT3对纤维化也有保护作用。Hexafluoro可在博来霉素暴露的小鼠中诱导SIRT3的表达,通过抑制TGF-β1降低胶原蛋白、纤连蛋白及α-平滑肌肌动蛋白(α-SMA)的表达,从而减轻肺纤维化[37]。此外,线粒体质量控制还依赖于酶泛素化的存在来清除受损的线粒体蛋白,该过程受到去泛素化酶(deubiquitinase,DUBs)的负调控,后者通过去除PARKIN添加的泛素标签来抑制线粒体自噬。USP30(ubiquitin-specific protease 30)是去泛素化酶家族中的一员,去泛素化USP30可以增强线粒体自噬,代表一种新的治疗靶点[38-39]。综上所述,针对线粒体质量控制的药理学调控以生物合成、线粒体自噬及分裂/融合为目标,从而恢复线粒体功能,最终预防肺纤维化的发生(表1)。
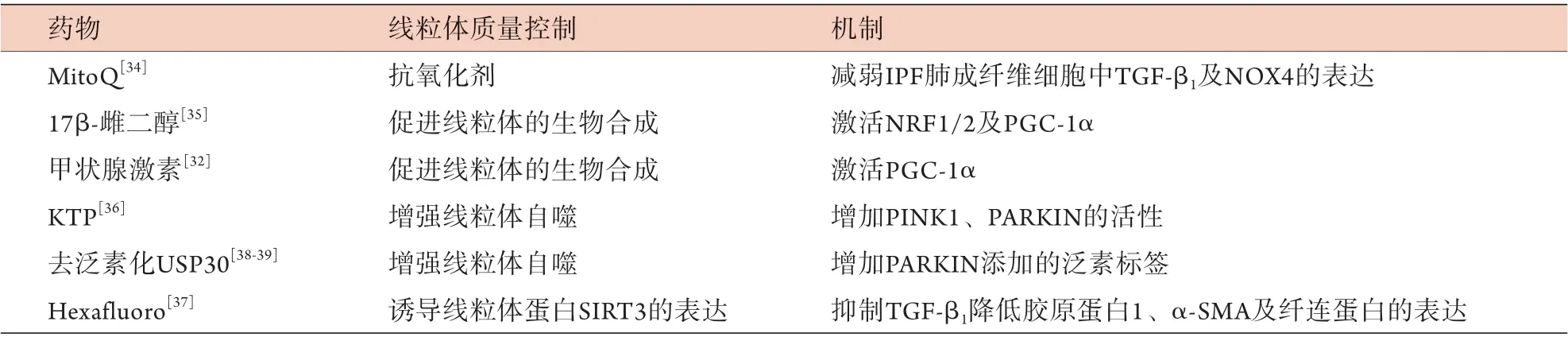
表1 线粒体质量控制干预IPF的现有潜在靶点Fig.1 Current potential targets for mitochondrial quality control in IPF intervention
4 总结与展望
在IPF背景下,线粒体质量控制失调导致线粒体功能障碍,ROS产生增加,诱发细胞凋亡。线粒体质量控制系统(线粒体自噬、线粒体生物合成及线粒体动力学)在IPF中起着不同的作用。IPF中线粒体自噬减少,受损的线粒体降解减少,功能障碍的线粒体聚积。IPF中线粒体的生物合成减少,无法补充新的线粒体成分,因而难以维持及恢复线粒体的数量与功能。TH对PGC-1α的上调作用逆转了博来霉素诱导的肺纤维化,这归因于线粒体生物合成增加,从而恢复了线粒体的功能。因此,在IPF中促进线粒体自噬及生物合成可能具有抗纤维化作用。线粒体融合及分裂不是两个独立的过程,而是两个相互依赖的过程。不同的压力应激环境能够决定线粒体动力学响应的方式。在轻度到中度应激环境下,线粒体融合将受损的线粒体与正常的线粒体结合起来以防止损伤,将多个线粒体融合为一个线粒体,使线粒体免于自噬。在严重的应激环境下,线粒体分裂将受损的线粒体区域与正常的线粒体分开,从而防止了进一步的损伤,受损的线粒体最终通过线粒体自噬去除。IPF中增强的线粒体动力学通过不断的融合与分裂,交换损伤的蛋白质及mtDNA,保持线粒体的动态平衡。
对于进行性发展的纤维化性肺疾病患者而言,目前仅有少量有限的有效治疗可供选择,需要进一步开展机制及转化医学等方面的研究。线粒体功能障碍引发纤维化反应的证据在IPF中尤为明显,但是将线粒体质量控制与肺纤维化异常修复联系起来的机制仍需进一步探索。
