基于飞秒激光诱导化学发光的流场速度测量研究
2022-04-01李明高强陈爽李博
李明,高强,陈爽,李博
(1 天津大学机械工程学院内燃机燃烧学国家重点实验室,天津300072)
(2 中国空气动力研究与发展中心,四川绵阳621000)
0 引言
气体流场的速度测量是流体科学中的一项重要任务,精确的速度测量有助于研究和了解与流动相关的量,如涡度和切应力[1-3],也有助于建立更加完善的数值模型,在研究湍流及热动领域有着十分重要的意义。传统的流场速度测量技术以侵入式测量为主,需要向待测流场中放置测量探针,常见的有皮托管测量法[4]和热线风速仪[5]测量法。传统的测量方法具有价格低廉的优点,但是会对流场产生干扰,而且测量探针还有一定的响应时间,影响测量的实时性和准确性。
激光测速技术属于非侵入式测量技术,对待测流场的干扰小,具有较高的测量精度,还可实现实时在位测量,所以近年来在流场和燃烧诊断领域得到了迅速的发展。根据测速原理的不同,激光测速技术主要分为两大类。一类是基于多普勒效应的测速技术,利用示踪粒子的散射光相对于入射光的频移来反演流场的速度信息,例如激光多普勒测速(Laser Doppler Velocimetry,LDV)技术[6-7]和平面多普勒测速(Planar Doppler Velocimetry,PDV)技术[8]。这类测速方法适用于高速流场,但速度反演的过程相对复杂,并且反演过程存在不确定性因素。另一类是基于示踪剂单位时间内的位移实现测速的技术,这类测速技术比较直观,能够实现对多点瞬时速度场的同时测量,还可获得与空间相关的信息。根据示踪剂的大小,这类测速技术又可分为粒子成像测速(Particle Image Velocimetry,PIV)技术[9-10]和分子示踪测速(Molecular Tagging Velocimetry,MTV)技术[11-12]。PIV 技术通过连续两次拍摄散布在流场中的示踪粒子,能够给出流场的二维速度信息,但在高速流场环境下,由于示踪剂粒子的跟随性较差,测量受到一定的限制。MTV 技术以分子作为示踪剂,不存在流场跟随性问题,大大提高了速度测量范围。MTV 技术经常以双乙酰[13]、丙酮[14]、二氧化氮[15]以及金属原子[16]等作为示踪分子,也可以直接标记流场中的分子,例如通过标记光解氧气产生的O3作为示踪分子的臭氧示踪测速(Ozone Tagging Velocimetry,OTV)技术[17];通过标记水蒸气解离产生的OH作为示踪分子的羟基示踪测速(Hydroxyl Tagging Velocimetry,HTV)技术[18-19];通过标记非线性受激拉曼过程产生的振动激发态的O2作为示踪分子的拉曼激发激光诱导电子荧光(Raman Excitation Plus Laser-Induced Electronic Fluorescence,RELIEF)示踪测速技术[20]和通过标记NO 作为示踪分子的空气光解-重组示踪(Air Photolysis and Recombination Tracking,APART)测速技术[21]。以上MTV 技术通常需要两束激光,一束激光标记流场中特定的示踪分子,另一束激光用来读取示踪分子,因此测量装置和测量过程比较复杂。
2011年,MICHAEL J B 等[22]开发了飞秒激光电子激发示踪(Femtosecond Laser Electronic Excitation Tagging,FLEET)测速技术,在纯N2流场中,通过飞秒激光光解N2产生的N 原子在重组过程中释放的长寿命荧光,实现对流场的标记测速。该技术只需一束激光便可实现速度测量,大大简化了实验装置和步骤。在此基础上,JIANG N 等[23]使用波长为202.5 nm 的飞秒激光,通过共振激发-光解过程实现对N2的标记,大大提高了FLEET 技术的激发效率。但总体而言,FLEET 技术的荧光信号强度仍然较弱,信噪比差,荧光寿命短,因此速度测量精度和测量范围具有一定的局限性,尤其是在低速流场环境下,由于需要长时间延迟(几十微秒延迟)才能获得可分辨的位移信息,此时荧光信号已极大衰减,导致该技术在低速条件下的测量效果较差。
针对此问题,本文开展了荧光信号增强方法研究,在N2中加入少量CH4,利用飞秒激光诱导CH4和N2发生光化学反应,产生发光强度强,持续时间长的氰基(CN)荧光信号,可以提高测速精度,扩大测速范围,相对于FLEET 测速技术更具优势。本工作主要研究该方法的荧光信号产生机制、增强效果和持续时间,进而分析该方法的测速范围、测量极限、测量误差以及延迟时间和激光能量对测速精度的影响等。
1 实验装置
本实验是在温度压力恒定的超净间内进行的,实验室温度20 ℃,湿度50 %,压力1 个大气压。实验装置如图1所示。本实验使用钛蓝宝石飞秒激光器(Spitfire Ace,Spectra-Physics)产生中心波长为800 nm 的飞秒激光,激光脉宽为45 fs,重频为1 kHz,最大脉冲能量为7 mJ。800 nm 的飞秒激光经过三倍频后输出中心波长约为267 nm 的飞秒激光。实验采用267 nm 的飞秒激光是因为相较于800 nm 的飞秒激光,267 nm 的飞秒激光诱导的光丝更细、更长,能够提高速度测量的精度,扩大速度测量的空间范围[24],同时267 nm 的飞秒激光所需的成丝阈值能量更低,有助于减小热效应等因素对流场的潜在扰动[24]。267 nm 的飞秒激光的最大脉冲能量为300 μJ,经过能量衰减器可以调节激光的能量。激光通过焦距为300 mm 的聚焦透镜聚焦于CH4/N2待测流场,产生飞秒光丝。CH4/N2待测流场由McKenna 燃烧器产生。CH4/N2混合气分别通过McKenna 的中心管和烧结多孔金属板产生中心射流和外围协流。中心管的直径为2 mm,烧结多孔金属板的直径为60 mm。气体的流速由质量流量控制器(HORIBA METRON)控制。
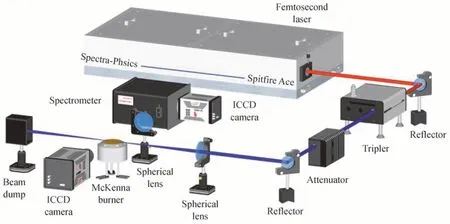
图1 实验装置Fig.1 Experimental setup
飞秒光丝在CH4/N2流场中会诱导化学反应,产生荧光信号。对于荧光光谱测量,使用焦距为100 mm的聚焦透镜,将荧光信号等大成像至光谱仪(Acton SP-2300i,Princeton Instrument)入射端的狭缝中。狭缝宽度为100 μm,平行于流场的流动方向放置,以获得沿流场速度方向的空间分辨光谱。光谱仪收集到的荧光信号经光栅(300 grooves/mm,闪耀波长为300 nm)分光后,由光谱仪出口端连接的ICCD 相机(PIMAX4:1024i,Princeton Instruments)拍摄成像。对于流场速度测量,其测量原理是激光标记流场中的分子,使其产生跃迁,发出荧光,经过一段延迟时间后,流场中的标记分子产生位移,基于位移和延迟时间,即可推算出流场的速度信息。被标记分子的荧光信号由一台ICCD 相机直接拍摄成像,以获得不同延迟时间下(微秒量级)被标记分子的位移。成像系统的空间分辨率为27 μm。
2 结果与讨论
2.1 光谱分析
为了确定荧光信号来源,实验在500 ppm(1 ppm=1×10-6mol/L)的CH4/N2混合气中进行空间分辨光谱测量。如图2(b)所示,CH4/N2混合气分别通过McKenna 燃烧器的中心射流管和外围多孔金属板产生中心射流场和外围协流场。图2(a)为飞秒光丝穿过流场激发产生的荧光信号,图中水平方向上的紫色细线为飞秒光丝所在位置,其激发的荧光信号随流场向上移动。从图中可以看出,该荧光信号非常明亮,且荧光信号的持续时间较长。图2(c)是荧光信号经光谱仪分光后,利用ICCD 相机成像获得的空间分辨光谱图,横坐标为波长,纵坐标为高度。ICCD 相机的延迟时间为0 μs,门宽为100 μs。沿着图2(c)中的白色虚线提取数据,可获得该位置处的光谱曲线,如图2(d)所示。从图2(d)中可以看出,较强的荧光信号分布在358 nm、387 nm、414 nm 和448 nm 附近。这四处信号都是由CN 自由基B2Σ+→X2Σ+的振动能级跃迁产生。
荧光持续时间决定了该方法的速度测量范围,本文研究了信号强度随延迟时间的变化关系。在CH4浓度分别为500 ppm、1 000 ppm、2 000 ppm 和5 000 ppm 的CH4/N2混合气中,使用ICCD 相机拍摄不同延迟时间下荧光信号的单脉冲图片,相机的门宽为1 μs,选取0.5~150 μs 范围内共11 个延迟时间点。将每张图片的荧光信号积分,得到整体荧光信号的强度,绘制的信号强度衰减曲线如图3所示。图3 的横坐标为延迟时间,纵坐标为信号的强度。从图3 可以看到CN 荧光的持续时间很长,可到微秒量级。而相关研究表明,CN的荧光寿命只有几纳秒[25]。原因在于飞秒激光将CH4/N2光解后,发生了持续产生CN 的化学反应。前期工作中,我们已经对持续的化学发光过程给出了相应的解释[26],如N+CH→CN(B)+H,生成直接处于高能级的CN(B)[27],或者处于低能级的CN(X)从N2(A)或N2(X)中获得能量并被激发到CN(B)态[28]。这些生成CN(B)的化学反应过程相对较慢,在微秒量级,所以CN 荧光的持续时间较长。而在荧光强度上,CH4/N2混合气流场的荧光信号要比纯N2流场的荧光信号强1~2 个数量级[26]。如此长的荧光持续时间和高强度的荧光信号可以为速度测量提供重要保证。
仔细观察图3 可以发现,信号强度随延迟时间呈先升高后下降的趋势。CH4浓度越高,曲线达到峰值的延迟时间越短,曲线的峰值越大,曲线的衰减速率越快。图3 中曲线先上升后下降是由于CN 的生成反应[27-28]和消耗反应[29-30]相竞争的结果,如CH4浓度为500 ppm 的曲线中,15 μs 之前CN(B)的生成反应占主导地位,CN(B)逐渐积累使信号强度增大,而在15 μs 之后,CN(B)的消耗占优势,信号强度开始下降。DILECCE G 等[31]在气体放电实验中,也观察到CN 荧光先上升后下降的趋势。图3 中CH4浓度越高,激光光解产生的含碳CN 前驱物越多,生成CN(B)的反应越迅速,所以曲线达到峰值所需的延迟时间越短,曲线的峰值越大。而在CH4浓度大于5 000 ppm 时,荧光寿命曲线呈现指数衰减。
由图3 可知,通过改变CH4的浓度可以改变荧光强度和荧光持续时间,而荧光强度直接影响速度测量的精度,荧光持续时间决定了速度测量的范围,所以在进行速度测量时,需要综合考虑测速所需的荧光强度和荧光持续时间,选择合适的CH4浓度,以实现最佳的速度测量效果。在测量高速流场时,几微秒的延迟时间便足够了,可以选择CH4浓度较大的CH4/N2混合气,在较短的延迟时间下获得更大的信号强度,从而提高速度测量的精度;而测量低速流场时,需要几十微秒甚至更长的延迟时间,可以选择较低的CH4浓度以获得更长的荧光持续时间。
2.2 速度测量
图4(a)和图4(b)分别为延迟时间0 μs 和10 μs 时,在2 000 ppm 的CH4/N2混合气中拍摄的单脉冲图片,相机门宽为1 μs,图片左上角为长度2 mm 的比例尺。外围协流气体的速度为0.1 m/s,在1 μs 的相机门宽内,位移可以忽略不计,所以认为延迟时间0 μs 时标记的协流分子的位置为测速起始时刻的位置,如图4(a)所示。在一段延迟时间内,标记的中心射流分子会发生位移,如图4(b)所示。将射流分子的位置减去协流分子的位置即为延迟时间内射流分子的位移。在进行测速实验时,由于光丝具有一定的直径,所以荧光信号也有一定的宽度,需要对荧光位置进行精确定位。分别截取图4(a)中的协流信号区域和图4(b)中的射流信号区域进行高斯拟合,取高斯拟合最大值所对应的位置为荧光信号的位置,如图4(a)和图4(b)中的红色虚线所示。图4(a)中的红色虚线为协流场荧光信号的位置,图4(b)中的红色虚线为射流场荧光信号的位置,两条虚线的距离为射流分子在延迟时间内的位移。由位移除以延迟时间,即可求出射流场的速度。
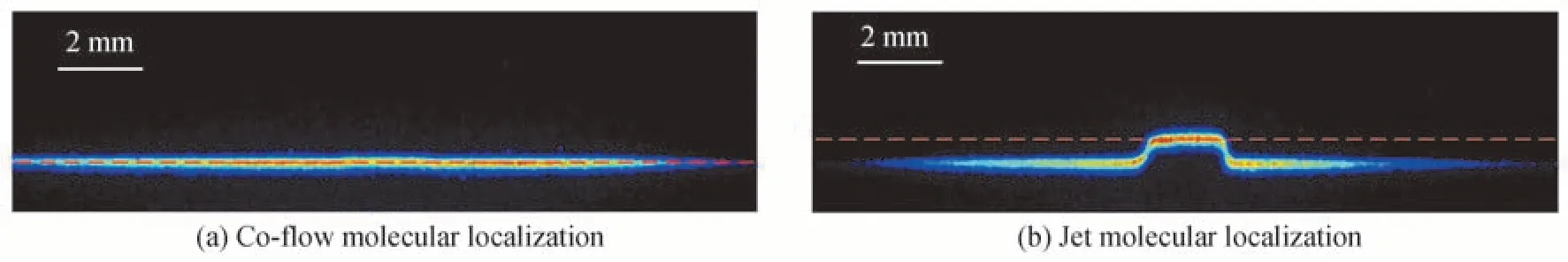
图4 标记分子定位示意图Fig.4 Location diagram of marker molecule
在进行速度测量时,荧光信号的宽度对测量精度具有一定的影响。宽度越窄,定位的准确度越高,速度测量的精度越高。荧光信号的宽度除了与光丝直径相关外,还受相机门宽的影响。相机的门宽越小,荧光信号的宽度也越小。但门宽过小又会使荧光信号强度减弱,影响定位的准确性,所以在进行速度测量时,需要综合考虑相机门宽对荧光信号宽度和强度的影响。在本实验的条件下,1 μs 的相机门宽最为合适,所以后续实验均选取1 μs 的相机门宽。
为了研究延迟时间和激光能量对测速的影响,选取不同的激光能量和延迟时间进行测速实验。图5 为不同的激光能量和延迟时间下,在2 000 ppm 的CH4/N2混合气中拍摄的单脉冲图片。图中射流场的速度为60 m/s,协流场的速度为0.1 m/s。激光能量依次为100 μJ、150 μJ、200 μJ 和250 μJ,延迟时间依次为2 μs、5 μs 和10 μs。每种工况下分别拍摄50 张图片,并对速度测量结果进行统计分析。
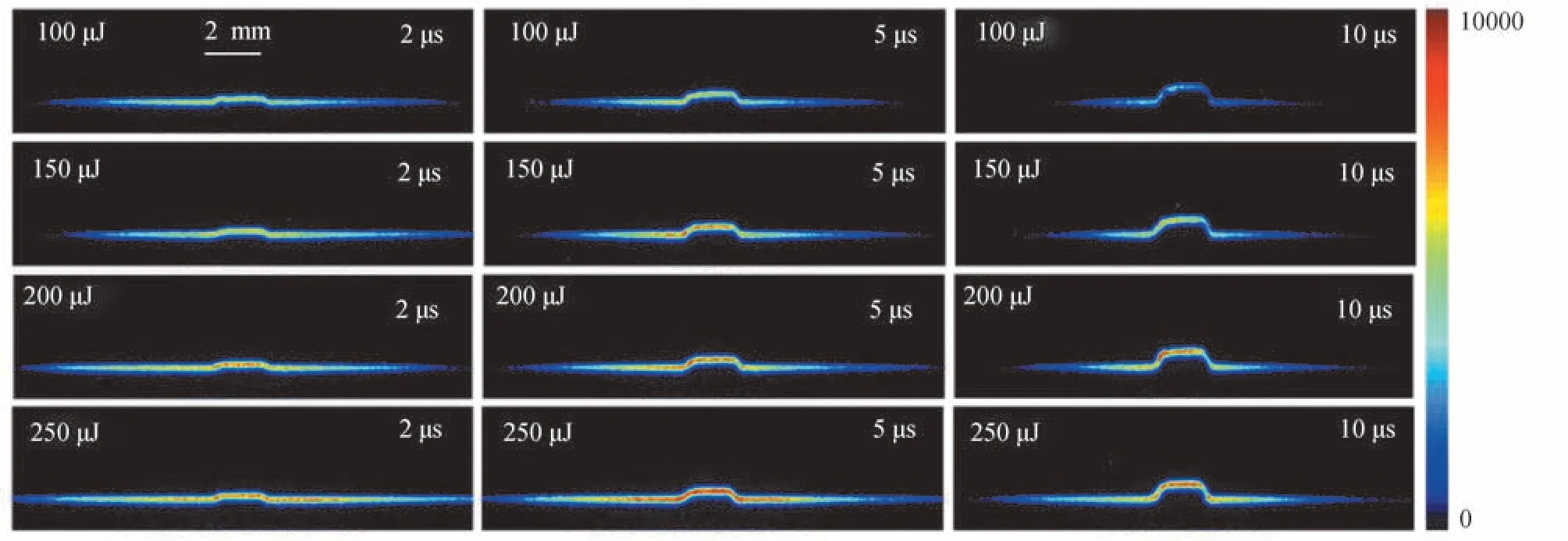
图5 不同激光能量和延迟时间的成像Fig.5 Images with different laser energies and delay times
从图6 可以看出,随着延迟时间的增大,整体速度呈现减小趋势。由于延迟时间增加,气体走过的距离变远,速度会逐渐降低,因此测得的平均速度也会减小。此外,随着延迟时间的增大,测量误差逐渐减小。由于该方法的测量误差主要来源于图像识别荧光信号移动距离S时产生的误差ΔS,而ΔS的大小只与荧光信号的宽度和强度有关,因此在延迟时间t增加时,ΔS/t会降低,使测量误差减小。如激光能量250 μJ,延迟时间2 μs 时的测量误差约为3%,而延迟时间10 μs 时的测量误差仅为0.8%。
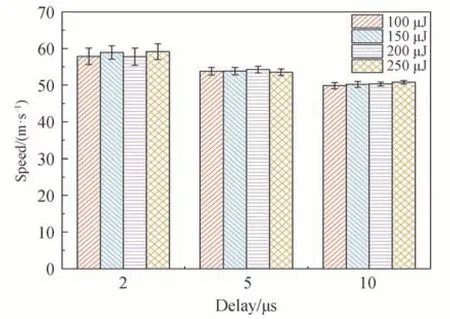
图6 不同激光能量和延迟时间的测速结果Fig.6 Velocity measurement results of different laser energy and delay time
从图6 还可以看出,延迟时间10 μs 时,随着激光能量的增大,测量误差呈现减小的趋势。结合图5 可知,10 μs 时荧光强度随着激光能量的增加而明显增强,使荧光定位的准确性提高,从而提高了测速精度。而延迟时间2 μs 和5 μs 时,在较低的能量下荧光强度已经很强,定位的准确性很高,继续增加激光能量对测速的精度影响不大。
本文还研究了该方法的速度测量范围。对于高速流场,流速越快,所需的延迟时间越短,荧光信号的强度越高,图片的信噪比(Signal-to-Noise Ratio,SNR)越大。在信号位移不变的情况下,延迟时间理论上可以设置为无穷小,因此本方法的测速上限理论上为无穷大。但实际测速上限还要受延迟触发设备硬件的时间分辨能力,成像系统的空间分辨能力和相机的最小门宽等因素的影响。对于低速流场,流速越低,测速所需的延迟时间越长,荧光信号的强度越小,图片的信噪比越差,所以本方法存在测速下限。
不同的CH4浓度,荧光信号的衰减速率也不同,需要选择合适的CH4浓度以实现低速流场的速度测量。图7 和图8 分别为不同延迟时间下,在2 000 ppm 和500 ppm 的CH4/N2混合气中拍摄的荧光单脉冲图片。图片左上角和右上角分别标有延迟时间和信噪比。为了便于观察,图片选取不同范围的色度条。对比图7 和图8 可以看出,CH4浓度为2 000 ppm 时,在50 μs 的延迟时间下图片的信噪比仅为3,而CH4浓度为500 ppm时,在120 μs 的延迟时间下图片的信噪比为8,说明通过调节CH4浓度确实能够有效延长荧光的持续时间,从而适用于低速流场的速度测量。图9 为信噪比随延迟时间的变化曲线。从图9 可以看出,500 ppm 的CH4/N2混合气在较大的延迟时间下,图片仍具有相对较高的信噪比,适合低速流场的速度测量,所以本文在该浓度条件下测量测速下限。图8 中500 ppm 的CH4/N2混合气在120 μs 的延迟时间下,图片的信噪比能够满足测速需求,所以保守认为120 μs 为该浓度下的最大延迟时间t。结合本实验中ICCD 的最小可识别空间位移S为27 μm,可以算出该浓度下的测速下限为0.23 m/s。进一步减小CH4浓度,可以获得更低的测速下限。
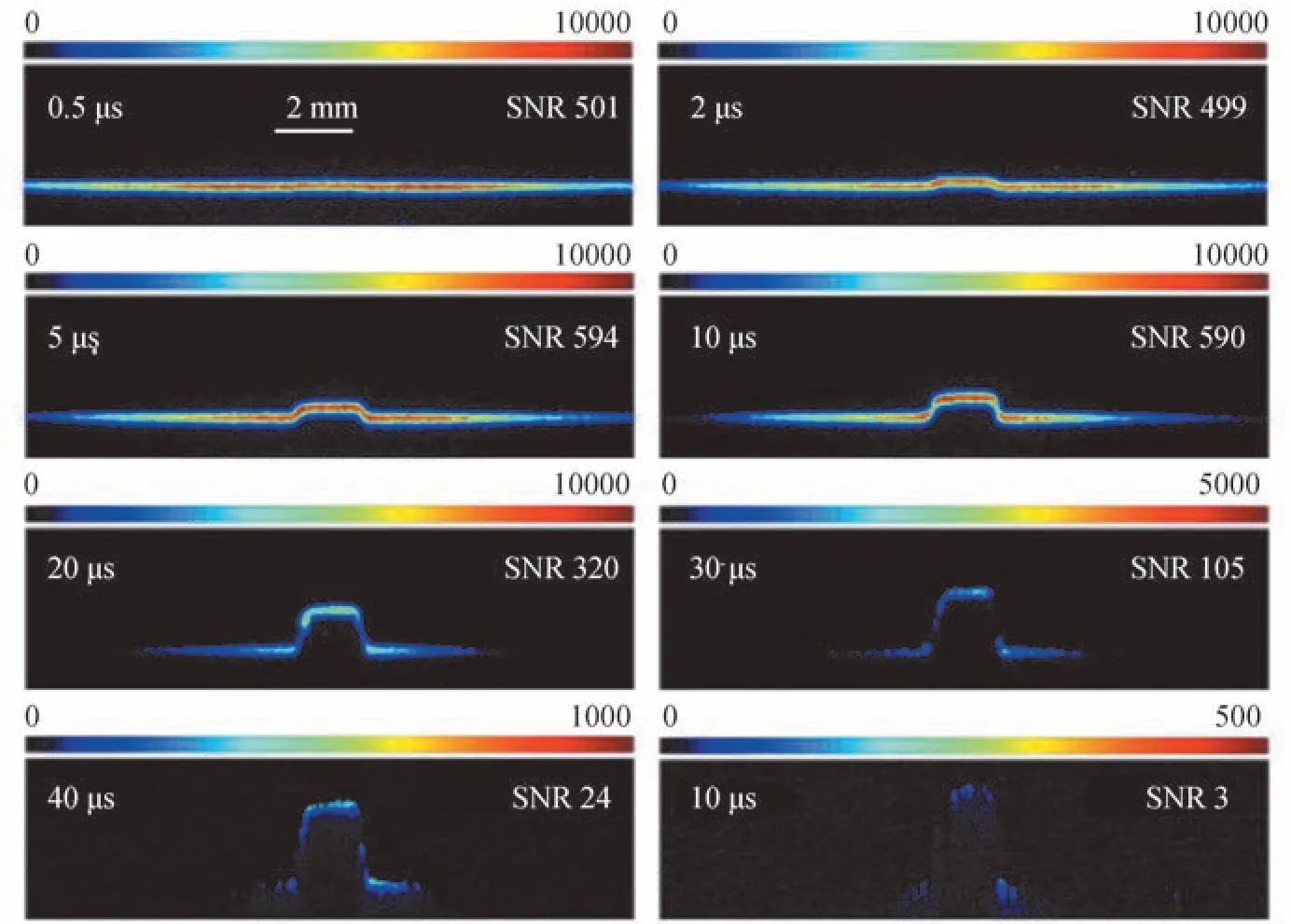
图7 2 000 ppm CH4信号强度随延迟时间的变化Fig.7 Signal intensity vs.delay times with CH4 concentration of 2 000 ppm

图8 500 ppm CH4信号强度随延迟时间的变化Fig.8 Signal intensity vs.delay times with CH4 concentration of 500 ppm
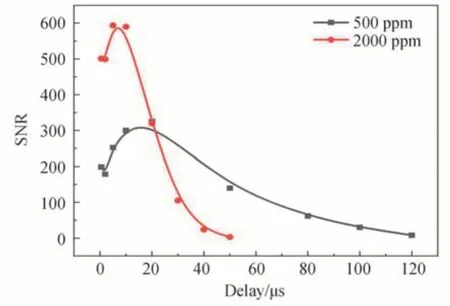
图9 不同CH4浓度信噪比随延迟时间的变化曲线Fig.9 Signal-to-noise ratio vs.delay time at different CH4 concentrations
本测速方法通过在N2中掺混少量CH4,实现了对荧光信号的增强和荧光持续时间的延长,相对于FLEET 测速技术更具优势。FLEET 技术的测速下限为7 m/s[32],而本方法大大扩展了测速范围,获得了0.23 m/s 的测速下限。由于本方法与FLEET 技术的测速原理相同,可以适用于所有满足FLEET 测速的场景,在超音速流场的速度测量方面有很好的应用潜力[22-33]。该方法也存在一定的局限性,例如本方法主要应用于一些非反应流场的速度测量,如超声速射流管,风洞等环境,在反应流场或燃烧场中还存在一定问题。
3 结论
本文研究了飞秒激光诱导化学发光测速的方法。利用267 nm 飞秒激光诱导CH4/N2混合气,生成发光强度强,持续时间长的CN 化学发光,并用于速度场测量。通过改变CH4浓度可以改变CN 荧光信号的强度和持续时间,进而适用于不同速度的流场,以获得最佳的测速效果。此外本文还研究了该方法的测速范围。该方法没有理论测速上限,但存在测速下限。在CH4浓度为500 ppm 的实验条件下,获得了0.23 m/s 的测速下限。与FLEET 技术相比,本方法信号强度提升了1-2 个数量级,信噪比高,提高了测速精度和测速范围,在流场速度测量中更具优势。本方法在航空航天等领域,如宽范围风洞速度场的测量与标定,具有很大的应用潜力。
