表面单分子量子态的探测和调控研究进展
2022-03-30姚杰赵爱迪
姚杰 赵爱迪
1) (中国科学技术大学化学物理系,合肥微尺度物质科学国家研究中心,合肥 230026)
2) (上海科技大学物质科学与技术学院,上海 201210)
1 引言
分子尺度的材料和结构由于其内禀的量子化能级结构、特殊的波函数特征,展示出了许多新颖的电子学、光学和磁学等物理性质,并可用来制备丰富功能的器件,如单分子开关、导线、整流器、晶体管、存储器和量子比特[1,2].无论是沿着经典比特的逻辑思路继续研究分子尺度的量子器件,还是开辟全新的以量子比特为基础的量子信息处理方式,都需要我们深刻理解分子尺度量子效应,发展新的量子设计方法与量子调控手段,构筑新的量子器件,最终实现信息的存储、传输与处理等多种功能.其中,单分子尺度结构的量子态的探测和调控是首当其冲的核心问题.在这里,量子态为广义的定义,即物质处于可由某一或若干量子数描述的状态,这些量子态可以被用于经典的非相干信息处理,也可以进一步被调控和用于量子相干信息处理.分子因其原子数目远小于固体,具有典型的分立的量子化能级,因此,对于分子尺度的物质而言,其量子态主要包括:分立能级的电子态(量子点的分立能级即与分子的类似);分立能级跃迁带来的光子态;局域磁矩带来的自旋态;其他的分立能态诸如振动态等也是量子化的.单分子尺度的量子态探测与调控的概念,起源于分子电子学研究热潮的兴起.在2000—2005 年左右,得益于扫描探针显微学的发展,使得对表面吸附单分子量子态的探测成为可能,涌现出单分子自旋态探测、单分子发光等一批重要的研究成果.经过十多年的发展,这一领域虽然已经取得了长足的进步,但也遇到了相当大的瓶颈,面临着诸多挑战,基于分子的量子器件的实现仍然道阻且长.问题诸如:已有分子不能满足实际应用的要求,需要对分子尺度量子结构进行新的设计和性能调控;基于分子的器件中分子与电极的连接不可或缺,所以需要深刻理解分子-电极之间、以及分子-环境之间的相互作用;分子量子器件在工作过程中必然伴随电荷与能量的转移,需要在分子尺度通过量子设计和调控,实现对相关电子态的激发、弛豫机制、寿命的准确了解和控制.在这篇综述中,我们将主要以表面单分子为对象,对其光子态、自旋态、电子态等的探测、表征与调控的相关研究做一下回顾,并提出一些下一步研究的设想,希望能抛砖引玉,寻找下一个突破口.
单分子量子态的探测和调控,首先面临的问题是研究手段的制约.对于单分子,通常附着于衬底,而扫描探针显微镜的发明,为探测单个分子带来了极大的契机.1981 年由IBM 的Binning 和Rohrer等[3]根据量子隧穿原理发明了第一台扫描隧道显微镜(scanning tunneling microscopy,STM)装置.1983 年,他们首次利用STM 观测到了Si(111)7×7重构表面的实空间原子成像[3].这也显示出了STM 极高原子分辨能力的优越性,这一发现将物质的微观状态以及原子和分子的实空间分布首次直观地展现在我们面前,使得原子和分子真实地被“看”到.之后在1990 年,IBM 实验室的Eigler 和Schweizer[4]在Ni(110)表面,利用STM 操纵35 个Xe 原子,将无序的Xe 原子排列成规整的IBM 这3 个字母,由此开创了STM 强大的原子操纵能力.这一工作也为之后的原子操纵构建全新的人造量子体系[5-8]以及人工精准调控单分子的化学反应打开了大门,激发了人们对STM 原子操纵能力的研究兴趣.另外在此基础上,STM 还开发出了电导谱学功能,结合STM 的高分辨性能可以探测分子体系局域的电学性能,主要包括I-V,dI/dV,以及d2I/d2V.I-V谱可以研究单分子的输运性质.而dI/dV谱以及图反映了电子结构和局域态密度分布,可以用来研究分子的电子结构.d2I/d2V谱是根据电子的非弹性隧穿原理得到的,可以用来研究化学键的振动模等信息[9,10],以及自旋激发信息[11,12].例如Yu 等[13]就利用dI/dV研究了分子轨道的纠缠态,而Ho 等[14,15]则利用d2I/d2V谱分辨出了单个分子的内部化学键结构以及分子间成键的化学键结构.为了进一步丰富STM 的探测范围以及提高STM 的分辨率,可以功能化STM 的针尖.例如在STM 尖端吸附1 个CO 分子,可以使得尖端更尖锐,提高STM 的分辨率,另外通过针尖吸附1 个磁性分子或者原子,可以实现对样品磁性自旋的探测[16-19].例如最近Czap 等[11,12]通过在STM针尖吸附1 个磁性的二茂镍分子,并用二茂镍分子功能化的STM 针尖再去扫描二茂镍单分子,实现了对二茂镍单分子自旋振动激发态和自旋交换相互作用的探测.
上述研究表明,STM 已经成为了目前研究表面单分子量子态最强有力的工具.除了STM 外,目前对于单分子量子态的探测手段还有非接触式的原子力显微镜(NC-AFM)[20-23]、太赫兹-扫描隧道显微镜(THz-STM)[24-28]、针尖增强拉曼光谱(TERS)[29-33]、STM 诱导发光技术(STML)[34-38]以及电子自旋共振(ESR)[39-41]等方法.而这些方法多在基于STM 技术上升级发展而来,需要与STM 一起联用.目前研究分子尺度的量子态主要以金属配位的酞菁分子和石墨烯纳米带这两类体系为载体,本文主要介绍基于STM 技术这两类体系中分子尺度量子态的探测和调控的研究进展,将先从表面单分子量子态体系的制备开始展开,再从单分子的光学性质和单分子的磁性自旋量子态以及最近兴起的石墨烯类分子的拓扑态和自旋态三方面重点介绍单分子体系的量子态探测和调控.
2 表面单分子量子态体系的制备
单分子体系的量子态可从制备、调控、表征三个方面进行介绍,如图1 所示,这里制备方法有STM 原位操纵和表面反应化学合成两种,单分子量子态整体可分为电子态[13,42]、光子态[43]及自旋态[44,45]三种,且对其单分子量子态的调控也主要针对这三种,其电子态、光子态、自旋态可以通过STM,THz-STM,NC-AFM,ESR-STM,TESRS,STML 等手段进行表征.化学合成的分子在自由空间中本身就有可能具有优越的量子特性,但当分子置于表面上后,其量子态的保持和探测往往并不容易,这其中最大的问题就来自于衬底与其相互作用会改变分子的量子特性.因此,表面单分子量子态体系的有效制备和获取是进一步探测和调控的前提.目前,制备合适的表面单分子量子态体系主要有原位操纵和表面化学合成两大路线,分别对应于传统材料的“自上而下”和“自下而上”制备方法.

图1 表面单分子结构的量子态的制备、表征与调控的研究示意图Fig.1.Schematic of the research on the synthesis,characterization and manipulation of the quantum states of surface-supported single molecular structures.
2.1 STM 原位操纵
STM 本质上是一种原位的近乎接触式的探测手段,可以就近实现对分子的操纵.因此,在STM发明后的十多年里,人们已经发展出了多种原位操纵的手段.比如机械操纵,通过控制针尖来拖曳或推动分子.最近的研究已经可以通过机械操纵的方法制备出多自旋耦合的人造单分子结构[46,47],演示了量子态的静态耦合.除了机械操纵外,通过针尖发射电子来“切割”和“焊接”分子中的特定基团,即电操纵,也已经被广泛用于实现对分子的改性,进而实现对具有可观测量子态的表面单分子结构的制备.2005 年,Zhao 等[48]利用针尖对吸附于Au(111)表面CoPc 分子进行“切割”,实现了其中Co 离子d 轨道中1/2 自旋态的恢复.CoPc 分子如图2(a)所示,从dI/dV谱上可看出吸附在Au(111)表面的钴酞菁分子没有近藤效应(图2(c)黑色线).用扫描隧道显微镜尖端的电压脉冲从分子上切下8 个氢原子(图2(b)),使该分子的4 个轨道与金基底发生化学键合.这种人工分子结构恢复了局域自旋,从dI/dV谱上可看出在费米表面附近观察到了明显的近藤共振(图2(c) d-CoPc)[48].通过这种方法首次实现了对单个分子自旋态的调控.类似的方法Li 等[49]也在FePc 上实现,利用分子手术切除了FePc 8 个瓣上的H 原子.不同的是,在切除之前,在dI/dV谱上观察到近藤共振,而切除最外层的8 个氢原子后,dI/dV谱特征变为双台阶结构,反映了分子自旋态的非弹性转变.

图2 (a) CoPc 的结构模型,在实验中,1 个瓣的氢原子2 和3 被解离;(b) STM 电流引起的脱氢示意图;(c) 不同温度下CoPc 和脱氢CoPc(d-CoPc)的dI/dV 谱;(d) STM 图像显示了连续尖端诱导的CoPc 在Au(111)上的脱氢[48]Fig.2.(a) Structural formula of the CoPc.Hydrogen atoms 2 and 3 of one lobe were dissociated in the experiments.(b) Diagram of the dehydrogenation induced by the STM current.(c) dI/dV spectra of CoPc and dehydrogenated CoPc (d-CoPc) at different temperatures.(d) STM images showing the sequential tip-induced dehydrogenation of a CoPc on Au(111)[48].
2.2 表面分子原位合成
STM 原位操纵虽然可以非常有目的地针对特定的局域化学键和基团进行操纵,但其效率很低,难以实现大规模的制备.2010 年左右,在表面直接合成分子(on-surface synthesis)的化学路线实现了重大的突破.Cai 等[50]利用“自下而上”的方法,通过小分子前驱体在金属表面反应辅助条件下合成了石墨烯纳米带.这种方法利用设计好的分子前驱体在金属单晶表面进行化学合成石墨烯分子结构,可以实现对石墨烯分子结构原子级别的精确控制.而且这种方法制备出的石墨烯分子结构质量高无杂质,有利于对石墨烯分子结构的性质探测.通过选取不同的前驱体,还可以合成不同类型的石墨烯分子结构.此方法中,首先通过设计和合成前驱体小分子,然后将前驱体小分子通过热蒸发的方式蒸镀到金属表面,例如Au(111)或Ag(111)表面.退火到一定温度,经过热活化后脱卤和乌尔曼偶联等反应后环化脱氢(cyclodehydrogenation)形成聚合的石墨烯分子纳米结构,用设计好的分子前驱体来进行精确控制“自下而上”地生长[51-58].图3 展示了合成具有不同手性石墨烯纳米带(chGNRs)的具体方法,包括所使用的前驱体小分子、合成路线,以及产物的STM 图像[59].

图3 结合溶液和表面合成chGNRs 的方法 (a)—(d) 溶液法合成不同原子宽度3,1,w-chGNRs 的分子前驱体1,2,3 和3,2,8-chGNRs 的前驱体4;(e)—(h) 利用4 种分子前驱体分别靶向chGNRs 的化学结构;(i)—(l) 在Au(111)表面合成chGNRs 的STM 图像[59]Fig.3.Synthetic strategy to produce chGNRs combining solution and on-surface synthesis:(a)—(d) Solution synthesis protocols for producing molecular precursors 1,2,3,for the synthesis of 3,1,w-chGNRs with different widths,and precursor 4 for 3,2,8-chGNRs;(e) —(h) targeted chemical structures of chGNRs by using the four molecular precursors in (a) —(d),respectively;(i)—(l) STM overview images of the chGNRs formed on a Au(111) surface[59].
3 单分子光子态的探测和表征
单分子光子态的探测和表征主要指利用单分子的量子特性使其作为单光子源的探测和表征研究.单分子具有分立的能级、确定的几何构型,这些与量子点都极为相似,而且单分子还天然具有全同性,使其发光特性具有极佳的一致性.单光子源毫无疑问是构建量子计算、量子密码、量子存储以及量子信息通信的核心[60-67].如何制备可控稳定高效的单光子源一直是量子光学追求的重要目标之一.相比于稀土离子[68,69]、量子点[70-72]、色心体系[73]的单光子源,单分子体系的单光子源发光频率丰富、量子产率高、室温操作且易于集成.早在2000 年Lounis 和Moerner[60]就利用脉冲激光激发固体中单个分子实现了可控的单光子源.这种方法触发单个光子产生的速率很高,与其他单光子源相比,具有简单、室温操作且可升级空间大的优势.因此单分子发光体系非常有潜力用来构建稳定可控且高效的单光子源.而基于STM 电流诱导的单分子发光,可以将STM 的空间分辨以及强大的对分子的操纵能力运用到诱导分子发光中,是到目前为止单分子发光研究的最主要手段.
在这里我们重点介绍运用扫描隧道显微镜实现的电致发光和光致发光.在用STM 对单分子进行发光研究的时候,一般常选择金、银、铜作为基本衬底,同时为了防止荧光淬灭,还需要让金属衬底和单分子脱耦合.实验中最常用的方法是在基本金属衬底上再通过热蒸发的方式生长NaCl 薄膜作为脱耦合的衬底,NaCl 薄膜的层数可以根据实验的需要自由控制.
3.1 单分子电致发光
目前对于单分子电致发光的研究主要有分为两种途径,一种是基于微纳加工,采用微纳电极直接对单分子施加电流作用从而使其发光[74].另一种是采用STM 技术,用隧穿电子诱导单分子发光[75,76].而鉴于STM 的实空间分辨能力以及其独特的可以对单分子进行操纵的能力,采用STM诱导的电致发光,更能从根本上研究单分子电致发光的机理.例如Svec 研究小组[77,78]采用CO 功能化STM 针尖后同时进行了针尖增强光子探测和高分辨率原子力成像,将NC-AFM,STM 及单分子电致发光有机结合,以及通过STM 对CuPc 分子的操纵,使其位于Na+或Cl—离子的中心,进而操控单个CuPc 分子的双激子态的能量变化.这些实验研究都展现了STM 对于研究单分子电致发光的独特优势.
电致发光机制中一个重要的现象为上转换发光.上转换电致发光是一种发射的光子能量高于激发电子能量的现象,其在激光信息技术、红外探测、生物医学等领域有巨大的应用前景.虽然在STM 隧道结内的上转换电致发光早期研究中已经提出了代表性机制,包括分子间三重态湮灭[79-81]和分子振动辅助等离激元泵浦[34,82].然而,早期的这些观察都是在分子多层膜上进行的,阻碍了竞争机制的潜在分化和主导机制的识别.2019 年Chen等[83]通过在银衬底上利用NaCl 薄膜作为脱耦合层,在其上沉积了酞菁分子,并结合STM 系统构建出酞菁单分子电致发光体系(如图4(d)).该工作在单分子水平上观测到电致上转换发光现象,有力排除了此前对这类体系中上转换机制的一些猜测.Qx(1.81 eV 单线态)峰值的强度在不同的偏压区呈现出3 个明显的逐步增加,表明不同的物理过程占主导地位.以此为基础的理论分析进一步表明,两种不同的分子激发机制——非弹性电子散射和载流子注入以及之间的微妙竞争导致了不同偏压区域不同的发光现象.基于对上述两种激发机制以及对系统其他潜在激发态的定量分析,首次提出了以自旋三重态作为中间过渡态,同时结合非弹性电子散射和载流子注入两种激发过程的单分子上转换发光机制[83].紧接着,他们又在多个单分子相干偶极体系中,发现了单光子的超辐射.在Ag(100)衬底上,利用NaCl 薄膜作为脱耦合层,同时利用STM 的单分子操纵能力,构建了由从2 到12 个非键合的锌酞菁分子链(如图4(a)),并且研究了这些分子链体系通过局域隧穿电子激发的发光特性以及随链长变化发光特性的演化[84].随着分子单体数目的增加,超辐射模式的峰位红移,且峰宽变窄.能量分辨的光子图可以反映出不同排布方式的偶极相互作用信息.对分子链的超辐射模式进行空间上的量子产率表征,可得到超辐射模式的光子图.共线同相的超辐射模式的能量分辨光子图表现出中间暗两边亮的哑铃形状图案(图4(b)).这种只有中间1 个节点的图案,说明Chen 等[84]构造的分子链在被电子激发后形成的是1 个耦合体系,测量的超辐射模式对应的离域激子态存在于整个分子链上.更为有意思的是,在分子链的辐射最强点测量了辐射光子的二阶相干函数,发现超辐射具有单光子特性(如图4(c)).这也证明整个分子链处于单激子态,必须被当作单一的量子体系来看待.当分子链体系中的1 个分子被局域电子激发后,激发能会迅速地离域到整个分子链,形成单激子超辐射态,进而产生单光子超辐射现象.该实验研究揭示了分子集体态的光学性质及其与分子尺度局域等离激元的相互作用.另外分子链超辐射模式的单光子辐射特性使得其可以作为单光子光源应用在量子计算和量子存储中[83].这一研究成果表明少数分子的聚集体也有望和单分子一样作为单量子体系用于量子光学等应用中.
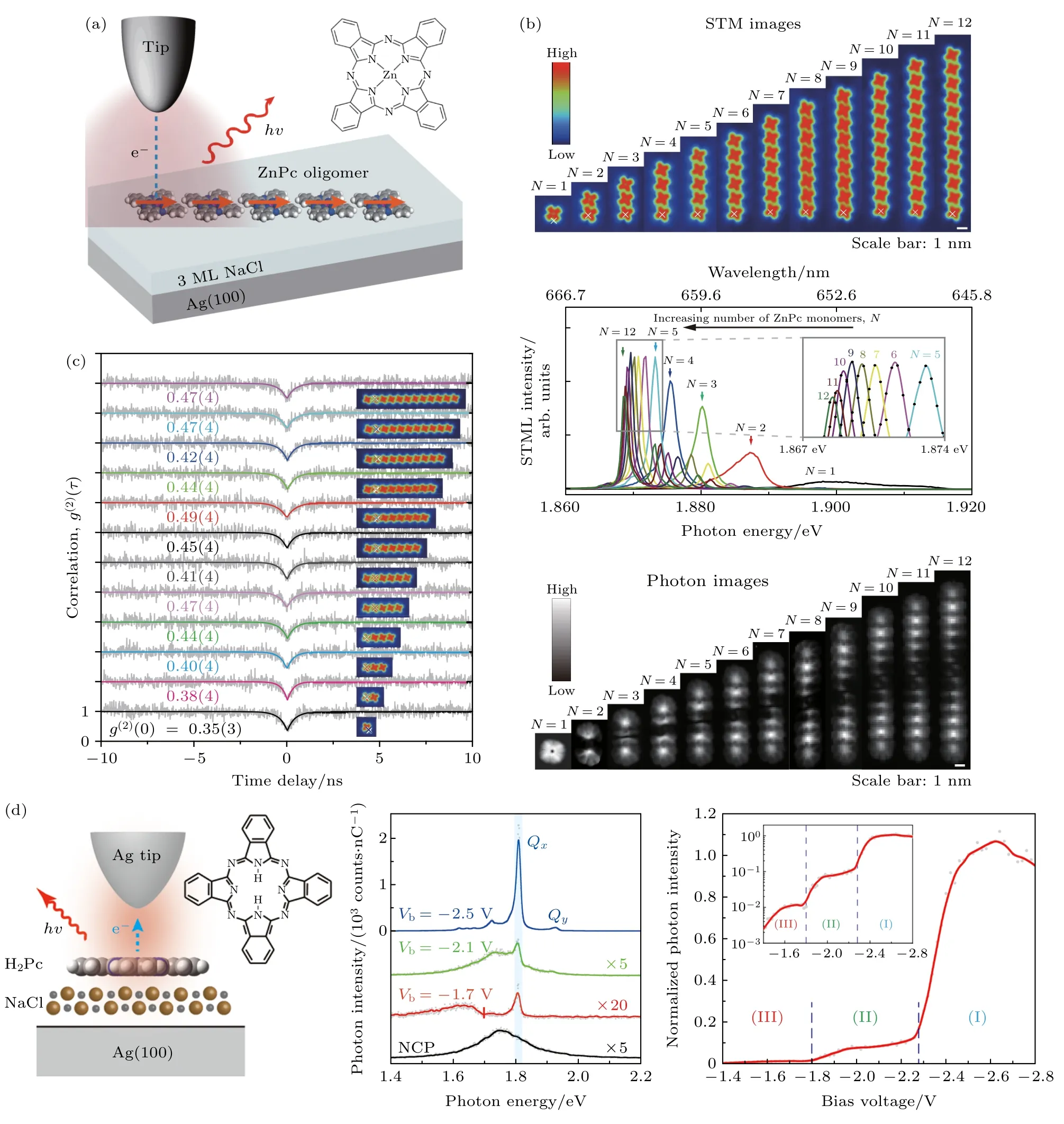
图4 (a) 诱导分子链发光的STML 示意图[84];(b) 多达12 个单体ZnPc 分子链的STM 形貌图,不同分子链的典型STML 谱,ZnPc 链超辐射态的实验光子图像[84];(c) 不同长度的分子链光子发射的二阶相关函数测量结果[84];(d) STM 诱导的单分子发光示意图,右边是分子结构的俯视图;不同偏置电压下单个H2Pc 分子的电致发光光谱;恒定电流下Qx 峰的归一化偏置电压和光强依赖关系图,对数图在插图中[83].Fig.4.(a) Schematic of STML from ZnPc molecular chains[84].(b) STM images of ZnPc molecular chains of up to 12 monomers,typical STML spectrum of different molecular chains,experimental photon images for the superradiant states of the ZnPc chains[84].(c) Second-order correlation functions g (2)(τ) for different ZnPc chains[84].(d) Schematic of the STM-induced single-molecule emission.A top view of the molecular structure is given on the right.Electroluminescence spectra from a single H2Pc molecule at different bias voltages.Normalized bias-dependent intensity of the Qx peak at a constant current,with the logarithmic plot shown in the inset[83].
3.2 单分子光致发光
尽管STM 诱导的单分子电致发光是一种独特而强大的研究光激发的技术,但它在激发过程中难以精确地选择性激发特定的分子能级.与光谱学中使用的可调谐激光器相比,隧穿电子的能量不是非常明确而且其能量也并非单色,这使得利用STM隧穿电流选择性地激发单个量子态变得特别困难.这阻碍了对单个激发态内在特征(如能级和线宽)的解释,以及对后续动态过程中由一种态到另一种态转变过程的描述.如果能利用STM 实现光致发光,将能使光致发光的分辨率提高到新的台阶.Yang 等[85]通过使用经过特殊修饰后的STM 针尖和金属衬底构造出1 个纳腔,并用激光激发其内部的等离激元场(图5(a)),高度局域的等离激元场使得单分子发射光子并且再次与等离激元场耦合至远场.ZnPc 单分子的光子图中呈现出明显的中间暗,而在4 个叶瓣上亮度有显著增强(图5(b)),这说明了电偶极模型的失效,需要将电子态跃迁密度也考虑进来.在光子图中沿着AB虚线做一条高度曲线,其中光子图的空间分辨率达到了惊人的8 Å(图5(c))[85].2021 年Imada 等[86]通过使用可调谐的激光代替固定能量的激光用于STM诱导单分子发光中(图5(d)),发现其发光强度得到了大幅提高.这种可调谐的单色纳米探测可以实现对单分子的单个电子和振动量子态的能级和线宽进行状态选择性表征.STM 的荧光光谱表明,纳腔等离激元的局部电磁场强烈增强光致荧光的过程,所有光致荧光峰均源自被测的单个分子.即使在非常弱的1 µW 激发功率下,总检测光子强度(能量积分)也超过180000 光子/秒,这表明其效率比尖端增强拉曼光谱和使用固定能量激光的光致荧光光谱高出几个数量级(如图5(e)).
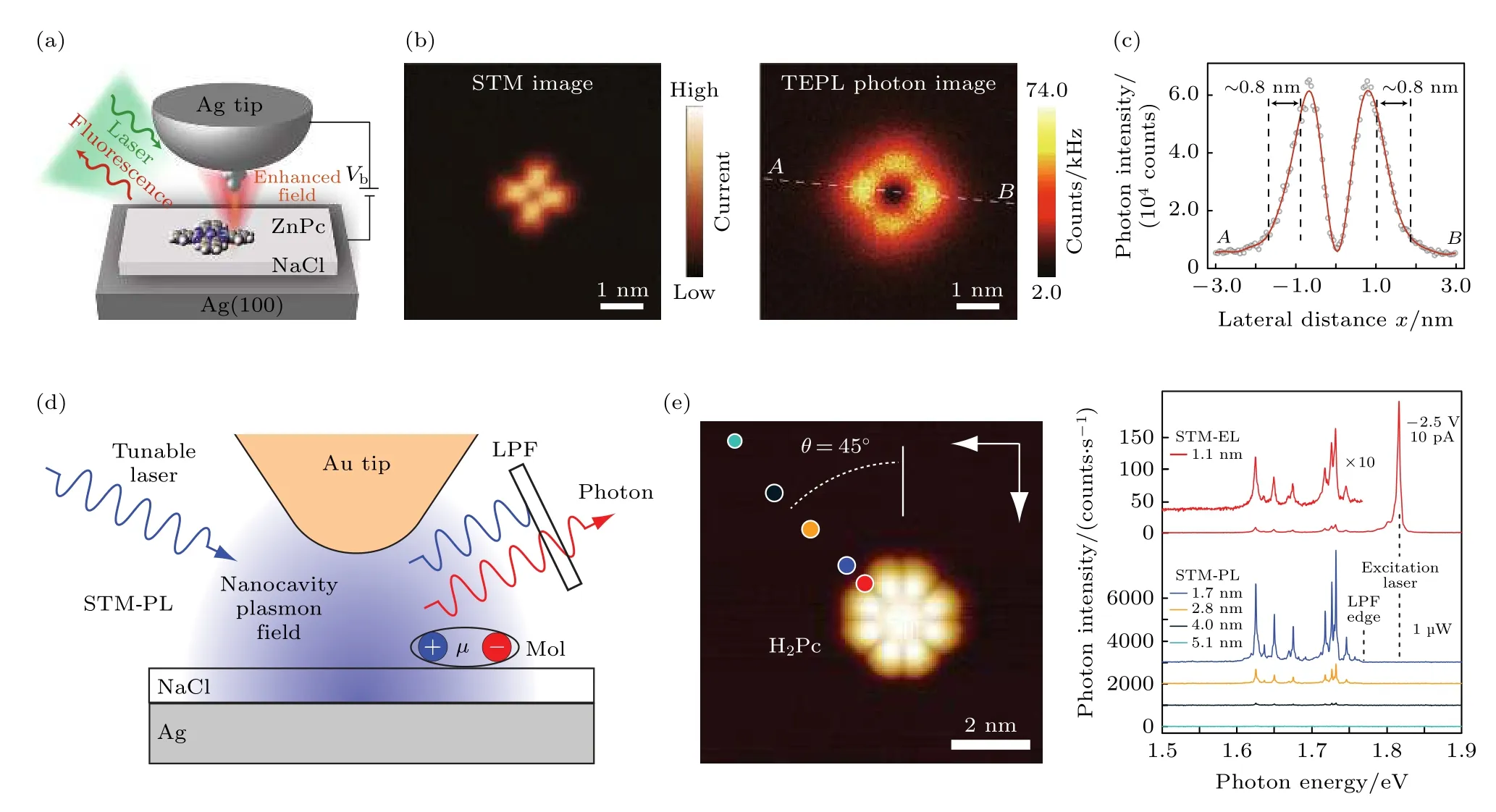
图5 (a) 针尖增强光致发光实验模型[85];(b) ZnPC 分子的STM 图(左)和光子图(右)[85];(c) 在光子图像(b)中虚白线AB 的光子强度侧面图[85];(d) STM-PL 实验示意图[86];(e) H2Pc 分子的放大 STM 图,STM 图(左)中的圆圈显示的是光谱测量时针尖的位置,其颜色与相应光谱的颜色匹配[86].Fig.5.(a) Schematic of the experimental of Sub-nanometre-resolved single-molecule TEPL[85];(b) simultaneously recorded STM image (left) and TEPL photon image (right) of a single ZnPc molecule [85];(c) photon intensity profile for the dashed white line AB in the photon image in b (right)[85];(d) schematic depiction of STM-PL measurement[86];(e) a magnified STM image of a H2Pc molecule,and the measurement tip positions for the spectra shown in STM image (left)are indicated with circles whose color matches that of the corresponding spectrum[86].
4 单分子局域自旋态的探测和调控
近年来单分子在纳米器件和自旋电子学中的潜在应用得到了广泛的研究.许多实验已经证明可以利用分子构成基本器件,包括负微分电阻隧道结[87]、整流器[88,89]、放大器[2]和数据存储[90].通过操纵单个磁性分子或原子的自旋自由度来进行对信息的编码是分子原子尺度电子学持续发展的核心挑战之一[91].而要想将其真正投入应用,那么关键的问题是对单分子的自旋态进行有效地控制.对表面单分子系统和分子水平的相关研究,不仅能从根本上理解分子系统的磁自旋特性,而且对于实现其实际应用具有特殊意义.尽管目前已经有多种检测和控制单分子自旋的方法,例如光学检测磁共振技术[92-94]、磁共振力显微镜[95-97]、氮-空位(NV)色心磁力计[98]和基于断裂结的分子装置[99]等.但这些系统的分辨率还不足以到单分子和单化学键的尺度,难以实现对分子内和分子间自旋-自旋相互作用的探测.STM 通过测量分子的微分电导(dI/dV)谱(即STS)来检测表面分子的局域磁性,可以实现精准地探测单分子间和单分子内部的局域自旋态.具体而言,对于表面吸附的磁性分子系统,STS 测量经常观察到近藤共振和自旋激发诱导非弹性电子隧穿的特征峰,并将其作为分析自旋信息的关键证据.近藤共振是费米能级附近的谱异常现象,起源于磁性分子的局域自旋与衬底传导电子之间的交换相互作用.因此,近藤效应通常在分子自旋载流子和导电衬底之间存在较强相互作用的系统中被观察到,如最近She 等[100]发现在半金属表面吸附的CoPc 分子中的单自旋能够得到很好的保持而不会淬灭.由于外部磁场或自旋-轨道相互作用,具有分裂自旋态的磁性分子,其自旋激发可以诱导非弹性电子隧穿,其dI/dV谱在费米能级处会表现出对称的阶梯状特征[44].另外通过STM 与电子自旋共振谱(ESR)的结合可以实现在成像的同时连贯地控制表面上单个原子的自旋.最近利用此技术研究了单配位配合物-铁酞菁(FePc),并研究了分子间自旋(FePc-FePc 二聚体)以及分子与原子间自旋(FePc-Ti)之间的磁性相互作用.并发现FePc 的分子自旋密度既分布于中心Fe 原子,又分布于配体(Pc),从而产生了强烈的分子几何结构依赖的交换耦合[39].在实现表面单分子局域自旋态制备和探测的基础上,如何对其进行调控是当前这一方向的研究重点.这其中,针尖作为不可或缺的一部分,可以通过施加局域电场来操纵分子的自旋态,也可以通过操纵原子实现自旋态的改变,还可以通过调控针尖物态和隧道结特性来实现对分子自旋态的调控.
4.1 针尖局域电场调控
2013 年,Liu 等[101]报道了一种可逆地调控单分子自旋态的方法.将MnPc 单分子吸附在Au(111)上,MnPc 分子表现出突出的交叉特征(图6(a)).通入H2导致原始MnPc 分子转化为中心有凹陷的结构(图6(b)).通入H2引起的MnPc 分子中Mn离子对H 原子的化学吸附,从而转化为H-MnPc.此外还发现,通过在H-MnPc 分子上施加正的尖端脉冲可以导致H 的分离,让H-MnPc 分子再次转化成MnPc 分子.H 原子对MnPc 的化学吸附也导致dI/dV测量值发生显著变化.MnPc 在Au(111)上的dI/dV谱在零偏压处呈现阶梯状特征(图6(c)中的红色曲线),且在施加磁场时可以使阶梯状特征发生分裂,因此可以推断出这种零偏压异常是由于近藤共振导致的(图6(d)).相比之下,H-MnPc 的dI/dV曲线无特征峰(图6(c)中的蓝色曲线).通过通入H2实现MnPc 到H-MnPc的转换,以及通过针尖脉冲使得H-MnPc 上脱H 重新变成MnPc,可以实现dI/dV谱上近藤特征曲线与无特征曲线之间的可逆转换(图6(c)).计算结果表明,与MnPc 相比,H-MnPc 中Mn 离子的d 电子数几乎不变,但有效电荷在d 轨道上重新分布,导致分子的净自旋从MnPc 的S=3/2 降低到H-MnPc 的S=1.此外,H 附着也导致Mn 与衬底更大程度地分离,从而削弱了Mn 与衬底耦合,这两个因素都有助于抑制H-MnPc 中的近藤效应[101].通过H 原子的化学吸附可以可逆调节MnPc 在Au(111)上的自旋态,这也为调控表面吸附单分子的自旋提供了思路,从而制备出具备量子自旋的单分子体系.

图6 Au(111)上 (a) MnPc 和 (b) H-MnPc 的STM 图像;(c) 由H 原子吸附和解吸引起的MnPc 分子中心记录的dI/dV 谱的连续变化;(d) MnPc 近藤特征在磁场下的演化[101]Fig.6.STM images of (a) MnPc and (b) H-MnPc on Au(111);(c) sequential variations of dI/dV spectra recorded at the center of a MnPc molecule induced by the adsorption and desorption of a H atom;(d) magnetic-field evolution of the Kondo feature of MnPc[101].
而同样利用针尖局域电场进行对分子磁性自旋的操纵,不同于Liu 等[101]的工作,Komeda 等[102]通过针尖脉冲控制分子旋转从而实现对自旋态的调控.dI/dV测量揭示了近藤效应特征的位点依赖性:近藤特征峰在分子的叶瓣周围很突出,但在中心处零偏压对应的dI/dV强度减弱(图7(a)).这表明近藤共振由Pc 配体上的自旋负责.然后,Komeda 等发现当通过对分子施加尖端脉冲来旋转上部Pc 配体时,可以实现可控地打开和关闭近藤效应.当TbPc2分子的2 个Pc 配体之间的方位角(θ)为 45° 时,其中心在STM 图像中显得更亮(图7(b)),可检测到近藤特征峰(图7(d) 中的顶部曲线).打脉冲后,分子的中心变暗,对应的TbPc2分子中2 个Pc 配体之间的方位角θ变化到了 30°(图7(b)).同时,dI/dV测量显示分子的近藤共振消失(图7(c) 中的底部曲线).近藤效应的这种依赖于夹角θ的转换被解释为分子自旋状态变化的结果.对于θ=45°的TbPc2分子,吸附在Au(111)上的分子的Pc 配体具有源自未配对π 电子的S=1/2 自旋.然而上配体旋转到θ=30°导致前沿分子轨道的重排,这使得电荷从表面转移,淬灭分子自旋,从而淬灭近藤状态.通过施加电流脉冲可以实现在2 个稳定的配体取向之间进行可逆切换,这能够实现在单分子水平上自旋态信息的编码[102].
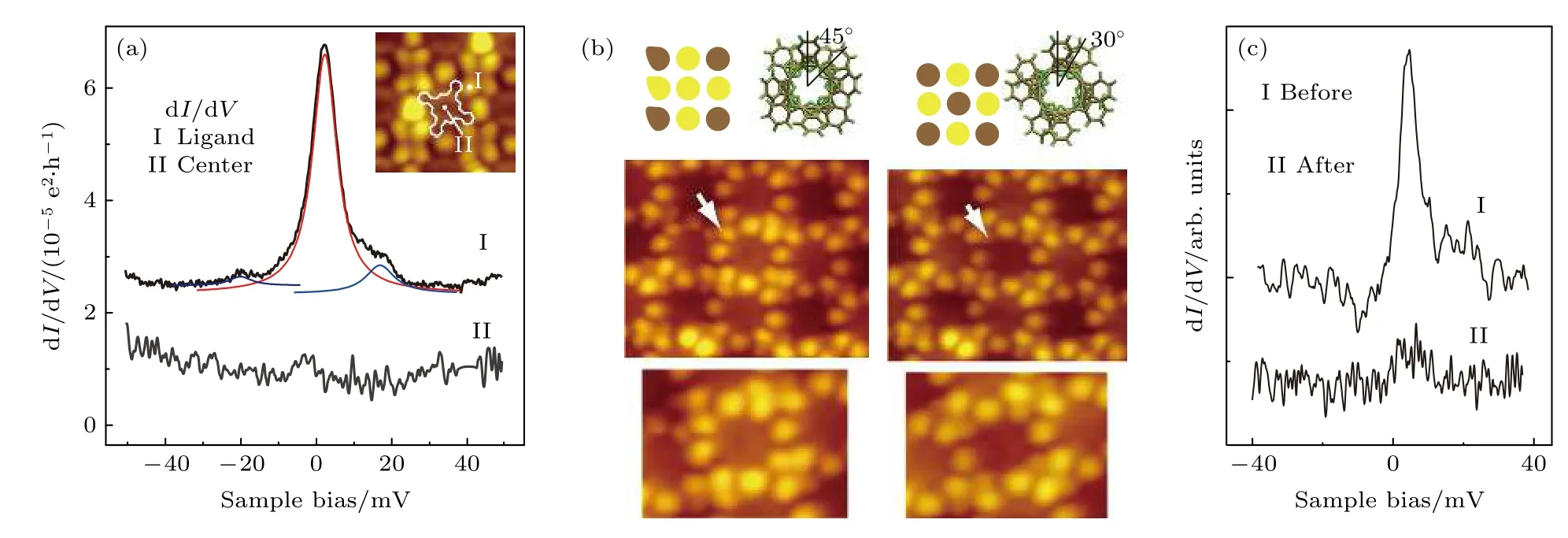
图7 (a) 在 Au(111) 上的 TbPc2 分子的叶瓣上和中心处记录的 dI/dV 谱.插图:组装结构中 TbPc2 分子的 STM 图像.(b) θ=45°和θ=30°的TbPc2 分子的示意图和STM 图像.(c) 在应用尖端脉冲之前和之后在 TbPc2 分子处获得的 dI/dV 谱[102].Fig.7.(a) dI/dV spectra recorded at the lobe and center of a TbPc2 molecule on Au(111).Inset:STM image of TbPc2 molecules in the assembled structure.(b) Schematic illustrations and STM images of TbPc2 molecules with θ=45° and θ=30°.(c) dI/dV spectra acquired at a TbPc2 molecule before and after the application of a tip pulse[102].
4.2 金属原子掺杂调控
虽然一些不含过渡金属的自由基分子本身即可以具有单自旋态[103],但如何能够调控一个无过渡金属的非自由基分子使其产生局域自旋是一个值得探索的方向.对于过渡金属参与的金属有机配合物,如金属酞菁(Pc)分子,因为d 电子的存在通常具有顺磁性.Pc 是一种广泛使用的大环螯合配体[104],Pc 与金属原子的配位通常是通过吲哚环的—NH—基团中的2 个H 原子的离解后发生的,导致Pc 配体的—2 价态.因此,稳定的金属酞菁(MPc,M=Mn[44],Fe[49],Co[48],Cu[105])分子更喜欢在Pc 和可以稳定在+2 价态的金属(例如3d 金属)之间形成.作为比较,无d 电子的主族金属Al(+3)并不易与PC 形成配合物.Hong 等[106]采用真空合成的方法,通过H2Pc 分子和Al 单质在 Au(111) 表面的共沉积,实现了对H2Pc 分子的金属化,制备出了AlPc 分子.图8(a)是H2Pc 和AlPc单分子的STM 图以及它们的结构模型.分别对这两种分子做dI/dV谱可以看出它们的电子态结构.在AlPc 叶瓣(图8(b)中的最顶部曲线)获得的dI/dV谱中检测到零偏置峰,证明了AlPc 中自旋的存在.该分子的自旋态密度起源于未配对π 电子,该电子主要在Pc 环的瓣上离域.这解释了AlPc中心没有近藤共振(图8(b)中的中间曲线).氯化的AlPc (ClAlPc)是一种由Al 原子与Cl 原子轴向键合形成的AlPc 衍生物,在Au(111)上的进一步比较研究(图8(c))表明,吸收Cl 原子后形成ClAlPc 分子没有近藤特征(图9(d)),因为氯化后ClAlPc 的所有分子轨道都被双重占据.因此,AlPc 在Au(111)上的自旋态可以通过Cl 原子与Al 中心的连接/分离来调节.这种方法也为调控无过渡金属分子的磁性提供了新的思路,即通过掺杂主族原子(如这里的Al 和Cl)实现对分子磁性自旋态的两级调控.

图8 (a) H2Pc 和Al 共沉积后的2 个分子的化学结构模型和STM 图.STM 图左上角和右下角的分子分别是H2Pc 和AlPc.(b) 将尖端置于AlPc 瓣(黑色)、AlPc 的Al 中心(蓝色)和H2Pc 瓣上方(黄色)得到的谱.(c) ClAlPc 的 STM 形貌图.(d) ClAlPc瓣的 dI/dV 谱.蓝色:Cl 向上,绿色:Cl 向下[106].Fig.8.(a) Chemical structure model and STM map of two molecules after co-deposition of H2Pc and Al.The top-left and bottomright molecules of the STM image are H2Pc and AlPc,respectively.(b) Spectra taken with the tip placed above a lobe of AlPc(black dots),the Al center of AlPc (blue),and a lobe of H2Pc (yellow).(c) STM topograph of ClAlPc.(d) dI/dV spectra of a lobe of ClAlPc.Blue:Cl-up,green:Cl-down[106].
4.3 隧道结调控
2017 年,Ormaza 等[107]通过在STM 金属针尖上吸附1 个二茂镍分子,构建出了金属针尖-单分子-金属衬底的单分子隧道结,可以实现单个分子自旋的可控转换.当改变从隧道区到接触区2 个电极之间的距离时,二茂镍分子从自旋S=1 可逆地切换到S=1/2.在可重复的电导测量中观察到的非弹性和弹性自旋翻转机制在实验上证明了这种切换,并通过第一性原理计算得到印证[107].
最近,Xing 等[108]也利用类似的方法构建出了单分子隧道结,在低温下通过简单地调节探针-样品距离来调节隧道耦合,实现了近藤共振峰和非弹性电子隧道谱之间的可逆切换.首先使用超导Nb 针尖区分吸附在 Au(111)上的 FePc 分子的两种不同构型(图9(a)和图9(b)).从形貌上看,它们表现出15°的旋转角度差,在用普通W 探针采集的微分电导(dI/dV)谱中,构型II FePc 在费米面附近出现急剧下降,这是由于其近藤效应导致.相比之下,构型I FePc 表现出在费米面附近出现一对对称台阶的特征,这些台阶特征来自于非弹性电子隧穿过程(图9(c)).利用超导Nb 针尖研究吸附在Au(111)上的FePc 分子,dI/dV谱显示了它们在费米能级附近都有1 个U 型能隙,该能隙归因于Nb 尖端的s 波超导能隙.同时除了针尖提供的额外超导能隙特征外,谱的台阶边缘也比W 针尖收集的更尖锐,这表明Nb 针尖的dI/dV谱能量分辨率得到增强(图9(d)).通过STM 针尖拾取1 个FePC 分子的方法,将隧穿结的结构从Nb 针尖-绝缘体-FePc-金衬底(图9 e)转变为Nb 针尖-FePc-绝缘体-金衬底结构(图9(f)),超导能隙内出现了类似Yu-Shiba-Rusinov 的特征(图9(f)),这是分子的磁性自旋态与Nb 针尖的超导态相互作用的典型特征.该研究表明,调整隧道势垒是一种有效的简单方法,可以在单分子隧道结中实现原位可逆的自旋开关[108].

图9 (a) 和 (b) 为组态I 和II 的 FePc 分子STM 图像,分别显示组态II 的“交叉”相对于组态I 的分子中心旋转15° ;(c) 通过正常W 端在构型I 和II FePc 分子上获得的dI/dV 谱,显示分子2 种构型的电子状态显著不同;(d)通过超导Nb 针尖在构型I 和II FePc 分子上获得的dI/dV 谱;(e) Nb-绝缘体-FePc-Au 隧道结结构以及其典型的dI/dV 谱,显示了Kondo 特征峰;(f) Nb-FePc-绝缘体-Au 隧道结结构,以及其典型的dI/dV 谱,显示了2 个间隙内YSR 态[108]Fig.9.(a) and (b) Typical STM images of configuration I and II FePc molecules,respectively,showing the “cross” of configuration II rotates with respect to the molecular center by 15° compared with configuration I;(c) dI/dV spectra obtained on configuration I and II FePc molecules by a normal W tip,showing strikingly different electron states for the two configurations of the molecule;(d) dI/dV spectra obtained on configuration I and II FePc molecules by a superconducting Nb tip;(e) typical dI/dV spectra in a Nb-insulator-FePc-Au tunneling junction,showing a Kondo dip;(f) typical dI/dV spectra in a Nb-FePc-insulator-Au tunneling junction,showing two in-gap YSR states[108].
5 石墨烯分子结构的量子态探测和调控
石墨烯是由排列成蜂窝状晶格的单层碳原子组成,由于晶格对称性,导带和价带在狄拉克点接触并具有线性色散关系.这一不寻常的电子结构使得石墨烯有望用于设计新范式特性的器件.最近兴起的转角双层石墨烯中关联电子效应的研究更是给石墨烯的电子学器件研究带来了新的方向.然而,单层石墨烯由于缺乏能隙,在电子器件中的应用受到限制.打开其带隙的有效方法是将石墨烯的横向尺寸减小到纳米级,变成石墨烯纳米带或者石墨烯纳米环等,通过量子限制和边缘效应引入带隙[109-111].这些石墨烯分子结构,本质上也可看成一种大分子,因此对其研究的方法和调控的手段也与单分子类似.石墨烯分子结构根据其边缘结构的不同,大致可分为扶手椅边缘和锯齿状边缘[112-114].其中扶手椅边缘的石墨烯分子结构表现出量子限制诱导的带隙,且其带隙依赖于带状宽度,这对其在电子学器件中的应用至关重要[115,116].相比之下,锯齿形边缘的石墨烯分子结构被预测具有自旋极化边缘态的未配对π 电子,其电子库仑斥力可以导致形成具有能隙的自旋极化电子结构.此外,这些锯齿状边缘的石墨烯分子结构还表现出几何和尺寸依赖的自旋输运性质[117,118].
在2.2 节中,我们可以看到,表面原位合成的方法已经使得制备各种不同宽度、不同异质结构、不同边缘结构的石墨烯分子结构成为可能,极大地拓展了单分子物态研究的范畴.近年来的研究表明,这些具有不同边缘、不同宽度、不同几何构型的石墨烯分子结构,可以表现出异常丰富的物性.其中最引人注目的是拓扑电子态和碳电子磁性.拓扑电子态本身并不一定是量子化的,然而对于拓扑绝缘体等体系的研究已经表明,拓扑电子态的实现和界面调控有望被用于实现量子化的局域态并用于量子计算.而石墨烯分子结构中的碳电子,则可以通过结构设计来实现无过渡金属原子的磁性,使得这些带有局域自旋态的石墨烯分子结构和磁性金属原子、NV 色心等单自旋体系一样有望用于量子信息,也有望用于自旋滤波器、自旋阀和自旋开关等自旋电子学应用[119].石墨烯分子结构的出现打开了石墨烯作为晶体管应用的大门,同时也丰富了石墨烯的用途,以及利用更加丰富的手段对石墨烯的各种物性进行调控[120].
5.1 石墨烯分子结构的拓扑电子态
随着石墨烯纳米带(GNR)拓扑边界态理论的提出,不同宽度、不同边缘和不同末端的半导体石墨烯纳米带属于不同的电子拓扑类.且石墨烯纳米带的拓扑相受空间对称性的保护,并由边缘的化学结构决定.不同拓扑相的2 个石墨烯纳米带之间的局域异质结电子态可以通过异质结边界几何结构来调节[121].如表1 所列,不同的边缘结构和不同宽度都会对拓扑不变数产生影响.这使得石墨烯纳米带的拓扑电子态具有很高的可调性,即通过化学方法设计和制备多样的几何结构,可以有效地实现对其拓扑电子态的调控.
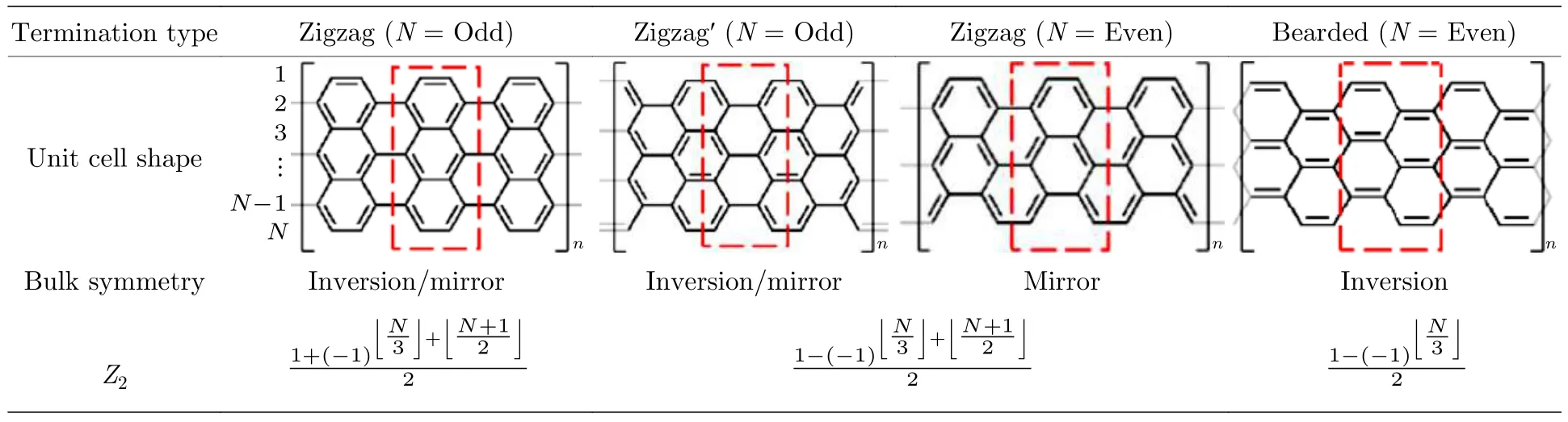
表1 AGNRs 的电子拓扑分类[121]Table 1.Categorization of electronic topology of AGNRs[121].
Gröning 等[122]在单晶表面上用小分子前驱体“自下而上”合成了所设计的石墨烯纳米带,并且通过理论模拟得出m≤ 3 时Z2=1,该模型构筑的石墨烯纳米带是拓扑非平庸的(图10(b)).在实验中利用小分子前驱体合成直型的具有扩展边缘异质结构超晶格的石墨烯纳米带,超晶格是由耦合的锯齿状小片段组成,并且利用STS 技术结合理论模拟证明了所构筑出来m=3 的石墨烯纳米带,在其导带和价带之间产生了拓扑边缘态(图10(c)).几乎在同时,Rizzo 等[123]预测出7—9 AGNR 超晶格将会在价带和导带间产生2 个能带,分别为占据的拓扑诱导能带和未占据的拓扑诱导能带(图11(b)).随后,他们利用了相同的合成原理,“自下而上”合成出了事先设计好的石墨烯纳米带,图11(a)是其化学结构模型以及不同结构的拓扑指数和高分辨率的STM 图像.这些石墨烯纳米带由拓扑不变数变化的节点周期性排列构成,从而导致具有不同拓扑不变数的石墨烯纳米片段异质结界面处拓扑态的形成.利用STS 谱学特征和理论计算模拟相结合(图11(c)),可以证明不同石墨烯分子结构异质结处的拓扑界面态以及石墨烯分子结构拓扑边缘态的存在[123].
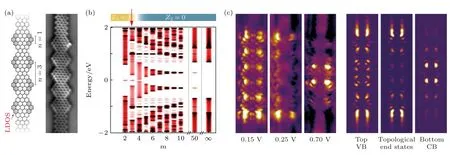
图10 直线型边缘扩展的AGNR 异质结构超晶格中的拓扑态 (a)化学结构和nc-AFM 图像;(b)计算的能带结构;(c)直线型边缘扩展 AGNR 异质结构超晶格的局域态密度(LDOS)图 [122]Fig.10.Topological states in in-line edge-extended AGNR heterostructure superlattices:(a) The chemical structure and nc-AFM image;(b) the calculated band structure;(c) the local density of states (LDOS) maps of an in-line edge-extended AGNR heterostructure superlattice[122].
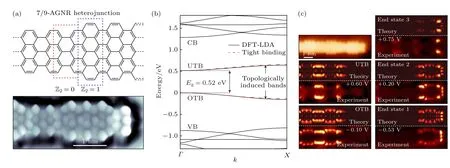
图11 7—9 AGNR 超晶格的拓扑态 (a) 化学结构模型以及不同结构的拓扑指数和高分辨率的STM 图像;(b) 计算的石墨烯纳米带能带结构;(c) 7—9 AGNR 超晶格的理论计算LDOS 图以及对应的实验dI/dV 图[123]Fig.11.Topological states in a 7—9 AGNR superlattice:(a) The chemical structure and high-resolution STM image;(b) the calculated band structure;(c) the LDOS maps and corresponding experimental dI/dV diagram of a 7—9 AGNR superlattice[123].
5.2 石墨烯分子结构的自旋量子态测量和调控
设计合成特定的石墨烯分子结构,还可以引入局域自旋,实现无过渡金属的仅由碳电子导致的磁性.磁性的引入可以通过设计特定形状和边缘结构,使其在π 电子的库仑斥力作用下自发形成磁性;或者通过局部引入五元环打破石墨烯分子结构的二分图对称性;或者通过引入像三角烯这样的自由基单元形成局域自旋.石墨烯分子结构中的自旋态具有显著的优越性.首先,通过合理地设计边缘或局域的化学键结构,可以精确地设计和调控石墨烯分子结构的磁性以及单自旋出现的空间位置,还可以设计多个自旋中心,并通过距离调控它们之间的相互耦合.其次,由于石墨烯分子结构全部由轻的碳元素组成,其自旋-轨道耦合和超精细相互作用极弱,有望具有较长的自旋态寿命和自旋扩散长度.
Li 等[124]通过设计小分子前驱体合成了具有3种不同异质结构拐点的石墨烯分子结构,如图12(a)所示,结合扫描隧道显微镜(STM)和扫描隧道显微谱(STS)技术,在1 型和2 型石墨烯分子结构中探测到了近藤效应(图12(b)和图12(c)),这表明在这些结构的拐角附近存在局域自旋.1 型结在PC 位置显示近藤效应,因为它有1 个H 原子结合到ZZ 位置,使磁矩猝灭,而2 型结则相反.3 型结在2 个位置是都无额外的H 原子饱和,因此S=0 整体不显示磁性,dI/dV谱上看不到近藤效应(图12(d)).此外实验中还使用了电子诱导脱氢让1 和2 型转变成3 型来调控石墨烯分子结构的磁性(图12(e)—(h)).该工作证明了石墨烯纳米条带拐角结构的π 电子磁性,并且深入揭示了与自由基位点结合的额外氢原子会淬灭它们的磁矩,从而改变所制备石墨烯分子结构的自旋态.

图12 (a) 3 种石墨烯纳米结的DFT 理论模拟.(b),(c) 分别为类型 1 和类型 2 结上明亮区域的近藤共振.零偏置峰值主要在类型 1 结的4 个 PC 环和类型 2 结的3 个 ZZ 环上被检测到.(d) 类型 3 结上零偏压附近的双峰特征.(e) 具有额外 H 原子2 个结的STM 图.(f) 在电子诱导去除额外的 H 原子后STM 图.(g),(h) 在脱氢过程之前(黑色)和之后(蓝色)的 PC1 和 ZZ2 区域的dI/dV 谱.(g)中的插图显示了脱氢过程中的电流变化[124]Fig.12.(a) DFT theoretical simulation of three graphene nanojunctions.(b),(c) Kondo resonances over the bright regions of Type 1 and Type 2 junctions,respectively.The zero-bias peaks are mostly detected over four PC rings of Type 1 junctions and over three ZZ rings of Type 2 junctions.(d) Double-peak features around zero bias over Type 3 junctions.(e) STM image of two junctions with extra H atoms.(f) STM image after the removal of extra H atoms induced by electrons.(g),(h) The PC1 and ZZ2 regions of the dI/dV spectrum before (black) and after (blue) the dehydrogenation process.The inset in (g) shows the current during the dehydrogenation process [124].
利用相似的合成方法,Mishra 等[125]在超高真空条件下Au(111)表面上用三角烯作为基本结构单元制备出了一维自旋链,其中三角烯是一种自旋为S=1 的二自由基多环芳烃.三角烯中的磁性源于其二分蜂窝晶格中固有的亚晶格不平衡而产生的,这使得其具有类似自由基的净自旋.三角烯及其衍生物难以通过传统溶液化学路线合成,但最近已在一系列金属和绝缘体表面上实现了原位合成,且其磁性可以在相对惰性的Au(111)表面上得以保持.因此,三角烯构成的自旋链(TSCs)为探索整数自旋S=1 链的拓扑物理提供一个理想的平台.在该实验中,通过使用STM 和STS 在原子尺度上探测了开放自旋链和闭合自旋环中与长度相关的磁激发,并直接观察了其中的自旋激发能隙和分数边缘态,如图13(a)和图13(b)所示为N=16 的开放三角烯链和闭合三角烯环的STM 图像,图13(c)为其上的dI/dV谱,可以看出只在开放三角烯链末端可观测到近藤效应的信号,表明其边缘态为S=1/2 的自旋态.图13(d)为理论计算给出的自旋链模型.计算展示了N=2—16 的开放三角烯链的自旋激发能,可以看出边缘激发能随N的增加呈指数下降(图13(e)).这一实验结果非常漂亮地验证了Haldane 早期的猜想,这是一种对称保护的一维拓扑链,其边缘S=1/2 的自旋态为无能隙的近自由态,而体态则具有能隙,与边缘导电、内部绝缘的拓扑绝缘体具有类似的拓扑物性.
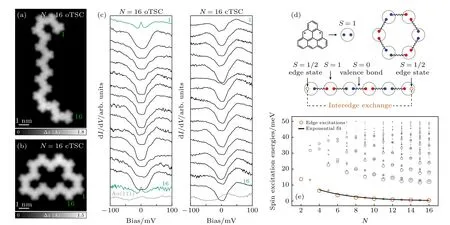
图13 (a),(b) N=16 开放三角烯链(oTSC)和闭合三角烯环(cTSC) 的高分辨率 STM 图;(c) 在N=16 oTSC 和cTSC 的每个单元上获得的dI/dV 谱;(d) N=6 oTSC 和cTSC 的价键固态自旋态,占oTSC 中S=1/2 边缘态,而cTSC 中没有;(e) 对于N=2—16 的oTSC,由BLBQ 模型ED 计算的自旋激发能量[125]Fig.13.(a),(b) High-resolution STM images of N=16 oTSC (a) and cTSC (b);(c) dI/dV spectra acquired on every unit of the N=16 oTSC (a) and cTSC (b);(d) the valence bond solid spin state for N=6 oTSC and cTSC,accounting for S=1/2 edge states in the oTSC and their absence in the cTSC;(e) For oTSC with N=2—16,the spin excitation energy calculated from the ED of the BLBQ model[125].
在具有纳米石墨烯结构的分子中引入五环结构,是实现全碳磁性的另一种手段.这主要是因为五元环的引入打破了正常石墨烯分子原本的二分图对称性,使得该单分子系统产生了自旋密度分布差和自旋极化.这样的石墨烯分子结构称为f-NG.一个典型的例子是Zheng 等[126]在金属衬底上精确合成了f-NG 及其共价偶联分子二聚体,并对其做了精细的STM 和STS 表征.在单个f-NG 分子上做dI/dV谱观察到零偏压处的近藤共振特征峰(图14(a),插图为f-NG 恒高的STM 图,数字位置对应于做dI/dV谱位置).峰值强度与空间自旋密度强度成正比.由于不对称的分子结构,共价偶联的f-NG 二聚体具有6 种不同的构型(标记为C1—C6),这取决于2 个嵌入的五元环的位置.2 个五元环位于每个f-NG (c3 和c4 构型)的同一侧,f-NG二聚体的基态为单线态,磁交换作用为3 meV.有趣的是,通过将2 个五元环相对放置(C5 和C6 构型),可以进一步增强交换相互作用,其中交换能量高达29 meV (图14(b)),这使得设计出室温的自旋电子器件成为可能.磁交换作用强度对嵌入五元环位置的强烈依赖性源于f-NG 单体内部不均匀的自旋密度分布.如图14(c)所示,通过实验研究了4 种不同的f-NG 二聚体,表示为C1—C4.对于C1,图14(d)中的dI/dV谱在2 个f-NG 单元上都显示出显著的近藤效应,表明2 个自旋之间没有强的磁耦合.对于C2 构型,五元环的位置与C1 相似,但2 个f-NG 单元相对于彼此倾斜,在2 个f-NG 长轴之间形成 133°的角.有趣的是,在C2 上dI/dV谱中观察到费米面两侧对称出现台阶,表明2 个自旋的反铁磁耦合具有2 meV 的磁交换相互作用.对于C3 和C4 结构,dI/dV谱表明2 个自旋也是反铁磁耦合的,但具有更大的磁交换能.这一实验结果证实了单个f-NG分子具有S=1/2 的单自旋,而在共价连接的f-NG 二聚体中,2 个单自旋之间可实现反铁磁耦合,其耦合强度随二聚体的连接形式不同而有所不同,这为调控f-NG 石墨烯分子结构的自旋提供了思路.
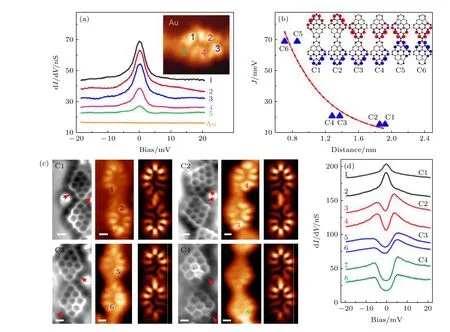
图14 (a) 在插图图像中彩色数字标记位置做的 dI/dV 谱.(b) 磁交换作用是每个单元中自旋密度最大的2 个碳原子之间距离的函数.插图为6 种不同f-NG 二聚体的自旋密度分布.所有二聚体都呈现单线态基态.蓝色和红色等表面表示自旋向上和自旋向下的密度.(c) 实验观察到4 种命名为 C1—C4 的 f-NG 二聚体构型.左侧,nc-AFM 图;中间,恒高的STM 图,右侧,模拟 STM图.(d) 在 (c) 中标记的位置做的 dI/dV 谱[126].Fig.14.(a) dI/dV spectra taken at the positions marked by colored numbers in the inset current image.(b) The magnetic exchange interaction J as a function of the distance between two carbon atoms with the strongest spin density in each unit.Inset:spin density distribution of six different f-NG dimers.All dimers exhibit a singlet ground state.Blue and red isosurfaces denote spin up and spin down density.(c) Experimental observed four configurations of f-NG dimers named as C1—C4.Left,nc-AFM frequency shift image;middle,constant-height current image;right,simulated STM image.(d) dI/dV spectra taken at the positions marked in (c)[126].
6 总结与展望
本文综述了基于STM 技术方向表面单分子量子态的制备、探测和调控的一些进展.我们可以看到,在技术方法上,STM 不仅可以实现对表面吸附单分子的空间高分辨和能量高分辨成像,还可以通过原位操纵以及结合外场和局域场等手段,实现对分子尺度的结构和电子态、自旋态等量子态的调控.除了直接操纵分子体系实现想要的人造分子结构,还可以通过STM 去操纵CO 分子构造出类似石墨烯的“反分子”人工晶格结构[127].STM 具备丰富的谱学方法,可以在表征单分子的隧道输运性质的同时,利用dI/dV谱和图像研究单分子体系局域态密度的空间分布,利用d2I/d2V二次微分谱,即非弹性电子隧穿谱(IETS),来研究单分子的振动激发和自旋激发.从STM 最早应用于单分子研究迄今,经过二十多年的发展,在STM 的基础上已经结合不同的技术,发展出了众多的联合探测手段用于原位的量子态表征,例如:STM-ESR,STMgate[128,129],THz-STM,STML,STM-SQUID[130],STM-磁场[131]等.
在体系上,随着表面原位合成技术的飞速发展和成熟,众多不同结构石墨烯类分子的设计、合成和制备都成为现实.人工设计定制的这些石墨烯纳米分子结构极大地丰富了表面单分子量子态的研究体系,其中拓扑电子态、局域自旋态的探测和表征已经展示出这一方向的丰富物理内涵.
在应用前景上,单分子发光对于构造单光子源以及信息传输有着重大的意义.分子天然具备有全同性,其发光频率易于调控且均一性好,并且分子可以通过修饰基团来提供极为丰富的调节手段.但现阶段半导体量子点的发光效率仍然更高并且制备更容易,单分子虽然有其优势,但难以封装,易受到外界干扰和影响等问题目前还没有较好的解决方案.单分子作为可实用化的量子光源还需要等待微纳技术的进一步发展.而表面单分子体系的拓扑物性、局域自旋等研究目前大多仍处于静态探测的阶段,要利用单分子体系的拓扑界面态、自旋态作为量子信息的载体,必须要解决来自衬底、分子环境等各方面对分子体系量子态相干性的破坏.而STM 作为一种近接触式的探测和表征手段,如何在探测自旋量子态的同时避免和消除这些退相干因素,有待新的方案的提出,和ESR 的结合能够部分解决探测的技术问题,但分子本身的隔离封装依然没有好的解决方案,目前使用的方法是在分子和金属衬底之间引入绝缘层,但这样隔离的分子难以在较高温度下稳定吸附.最近对二维材料的研究或许可以带来一些启发.在二维材料的器件中,为了最大程度减少对二维材料性质的影响,常用的绝缘层和封装层不再是硅基半导体常用的氧化物,而是使用单层六角氮化硼作为隔绝层.氮化硼非磁性且绝缘性极好,化学性质为惰性,与二维材料几乎没有电子和自旋相互作用.同样地,如果能够用氮化硼作为封装隔绝材料,把单分子量子体系封装在两层氮化硼中,那么单分子的电子学性质、自旋性质、光学性质的量子特性是否有望像金刚石中的NV 色心一样得到很好的保持,这需要实验和理论的检验.我们期待这一领域下一阶段重要的突破来自于分子体系量子态的有效封装,可以使得单分子相干量子态的研究成为可能,从而实现对单分子体系优越量子特性的真正利用,为基于单分子量子态的量子信息提供物质载体.
