紫花苜蓿根际土壤细菌群落对腐熟牛粪响应
2022-03-28李文辰赵金波张智慧
王 芳, 李 伟, 刘 鑫, 李文辰, 赵金波, 张智慧, 杨 曌
(1.齐齐哈尔大学生命科学与农林学院, 抗性基因工程与寒地生物多样性保护黑龙江省重点实验室, 黑龙江 齐齐哈尔 161006;2. 黑龙江省农业科学院畜牧兽医分院, 黑龙江 齐齐哈尔 161005; 3.黑龙江省牧草育种与种质资源利用工程技术研究中心,黑龙江 齐齐哈尔 161005)
微生物是土壤生态系统中的重要组成部分,驱动着土壤系统中各种生物进程。微生物对环境变化敏感,施肥显著影响土壤微生物种群特性,长期施用氮、磷、钾化肥可降低土壤微生物活性及多样性[1]。添加粪肥、植物残体等有机肥可改善土壤肥力、提高土壤有机质含量,稳定土壤微生物系统[2-4]。不同养分含量的有机肥对土壤微生物主要类群的影响不同。新鲜猪粪或发酵猪粪可以明显提高水稻(Oryzasativa)根际土壤微生物的物种丰富度和群落均匀度,加快有机物质在土壤中的分解转化[5]。蚯蚓粪会增加土壤有益微生物种类和数量,提高土壤细菌多样性,增强土壤酶活[6],对番茄(Solanumlycopersicum)连作土壤细菌群落结构的调节效果优于稻草、鸡粪、牛粪有机物料[7],但有研究表明牛粪可以对放线菌优势群落产生显著影响[8]。
土壤细菌作为土壤肥力和养分循环的驱动者,在维持农业生态系统的生产力和稳定性方面具有不可替代的地位[9-10]。细菌群落对土壤条件包括水分、温度、pH值、土壤类型和养分有效性等变化的反应快于其它化学或物理性质的反应[11],细菌群落结构和多样性可以作为评估农业措施对土壤质量影响的重要指标[12-13]。研究发现有机肥处理的土壤中细菌的优势种群明显不同于单施化肥和不施肥[14]。
紫花苜蓿(Medicagosativa)为多年生豆科植物,含有丰富的营养成分,被誉为“牧草之王”,因其较高的营养价值和较强的地域适应性,在我国北方畜牧业发展中具有举足轻重的地位。为了满足苜蓿正常生长对养分的需求,常需通过施肥来补充土壤养分。牛粪是畜牧产业的废弃物,将其作为有机肥料用于作物种植,形成种养有机结合的循环模式,不但解决了粪便污染和化肥过量使用的问题,而且可以改良土壤、培肥地力、提高作物产量及品质,实现经济及生态效益双丰收。本研究基于16S rRNA基因高通量测序技术,探索不同添加比例腐熟牛粪对苜蓿根际土壤细菌丰度、多样性及群落结构组成的影响,旨在为腐熟牛粪在农业中的应用及施肥策略制定提供科学依据。
1 材料与方法
1.1 试验设计与样品采集
试验于2020年 4月至9月进行。供试紫花苜蓿品种为‘龙牧801’,土壤采自黑龙江农业科学院畜牧兽医科研试验地(齐齐哈尔市富拉尔基区)。采用盆栽(盆钵25 cm ×30 cm),每盆均播种50粒种子。4种质量百分比添加腐熟牛粪,① O组(零添加组,0% 添加量);② D1组(3% 添加量);③ D2组(6% 添加量);④ D3组(9% 添加量),分别在播种后(苜蓿分枝期)的30 d(T1),60 d(T2),90 d(T3)取样,共12个处理,每个处理5次重复。此外,在添加牛粪前,取全土(不添加牛粪,WT)和全粪(不添加土壤,WF)两个样品,以了解未经处理及种植的土壤及腐熟牛粪细菌群落情况。采用抖落法收集苜蓿根际土壤,混匀后分装在聚乙烯袋中,-80℃保存,作为总DNA的提取样本。
1.2 土壤总DNA提取、PCR扩增及基因测序
每个样品称取0.5 g,根据Mo BIO Power Soil DNA Kit说明书提取总DNA,1% 琼脂糖凝胶电泳检测DNA完整性。以稀释后的基因组DNA为PCR模板,利用细菌通用性引物338F(5′-ACTCCTACGGGAGGCAGCA-3′),806R(5′-GGACTACHVGGGTWTCTAAT-3′)扩增细菌16S rRNA V3-V4可变区。扩增体系为 25 μL,5×FastPfu缓冲液 4 μL,2 μL 2.5 mmol·L-1dNTPs,0.8 μL引物(5 μmol·L-1),0.4 μL FastPfu聚合酶;10 ng DNA模板。PCR扩增程序为:95℃预变性 2 min,30 个循环(95℃变性 30 s,55℃退火 30 s,72℃延伸 30 s),最后 72℃延伸 10 min。PCR产物经1.2% 的琼脂糖凝胶电泳,利用SanPrep柱式DNA胶回收试剂盒(生工生物工程,上海)进行纯化。获得PCR产物送至派森诺生物科技有限公司(上海)进行Illumina-Miseq高通量测序。原始数据提交至NCBI的Sequence Read Archive (SAR)数据库,Accession号码:PRJNA741810。
1.3 原始数据处理
采用DADA2方法[15]对序列进行质控、去噪(Denoise)、拼接和去嵌合体,提高后续序列融合比率。利用Vsearch方法[16]聚类获得操作分类单元(Operational taxonomic units,OTU),统计长度分布情况,检查目的片段长度范围及异常长度序列情况。随机抽取序列,构建稀释性曲线。
1.4 物种分类学注释及物种组成分析
利用QIIME2软件,选用Greengenes数据库(http:// greengenes.secondgenome.com/)进行细菌16S rRNA基因注释。使用QIIME2(2019.4)及自编perl脚本,依据序列物种分类学注释的结果以及选择的样品,统计这些样本的物种注释结果中域、门、纲、目、科、属、种七个分类水平各自含有的分类单元数量,用柱状图呈现分析结果。
1.5 Alpha多样性分析
利用派森诺基因云平台(https://www.genescloud.cn/)进行OTU丰度和Alpha多样分析,Chao1和Observed species指数表征丰富度,Shannon和Simpson指数表征多样性。
1.6 Beta多样性分析
使用抽平后OTU表,基于Bray-Curtis距离矩阵进行PCoA(Principal coordinate analysis)分析,用QIIME2使分析结果可视化,R脚本输出样本点的PCoA坐标,绘制二维散点图。
1.7 数据统计分析及制图
使用IBM SPSS Statistics 19.0 统计分析软件对Alpha多样性指标、样本多样性数据进行二因素方差分析(WF和WT两个样本不参加方差分析),多重比较采用Duncan检验法,差异显著性水平为P<0.05。
2 结果与分析
2.1 测序数据分析
将测序平台得到的原始数据去除低质量、去噪、去嵌合体、拼接后,供试样本共获得984 200条高质量序列,去除singleton后的序列量是942 003条,总碱基数为392 582 624 bp,碱基数分布在403~442 bp的序列占序列总数的99.98%,平均序列长度为 417 bp。14个样品的稀释性曲线均趋于平坦(图1),表明测序数据接近饱和,测序深度合理,更多的数据量对发现新的OTU贡献率较小。同时,稀释曲线在一定程度上也可以反应在一系列给定的测序深度下,可能包含的物种总数及其中每个物种的相对丰度。在相同的测序深度下,比较不同样本中OTU数的多少,在一定程度上衡量每个样本的多样性高低。由图1所示,WT样本的多样性最高,其次为O组、D1,D2,D3组,WF样本的多样性最低。
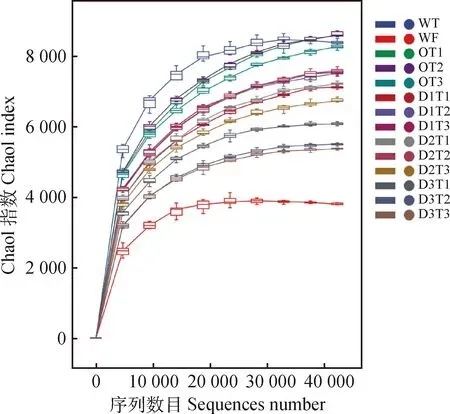
图1 样品稀释曲线Fig.1 Rarefaction curves for samples
2.2 物种分类学注释
将获得OTU从门到种进行物种分类学注释分析。所有供试土壤样品共注释到细菌分类85门,335纲,362目,1 828科,4 441属,368种。样品WF在门和属水平上注释到的OTU数目在所有样品中均为最低,分别为16和1 807,在种水平上为最高368个。WT在门水平上注释到的85个OTU,在所有样品中为最高。在属水平上,OT3注释到的OTU数目最多,为4 441个;在种水平上注释到的OTU数目为最低182个(图2)
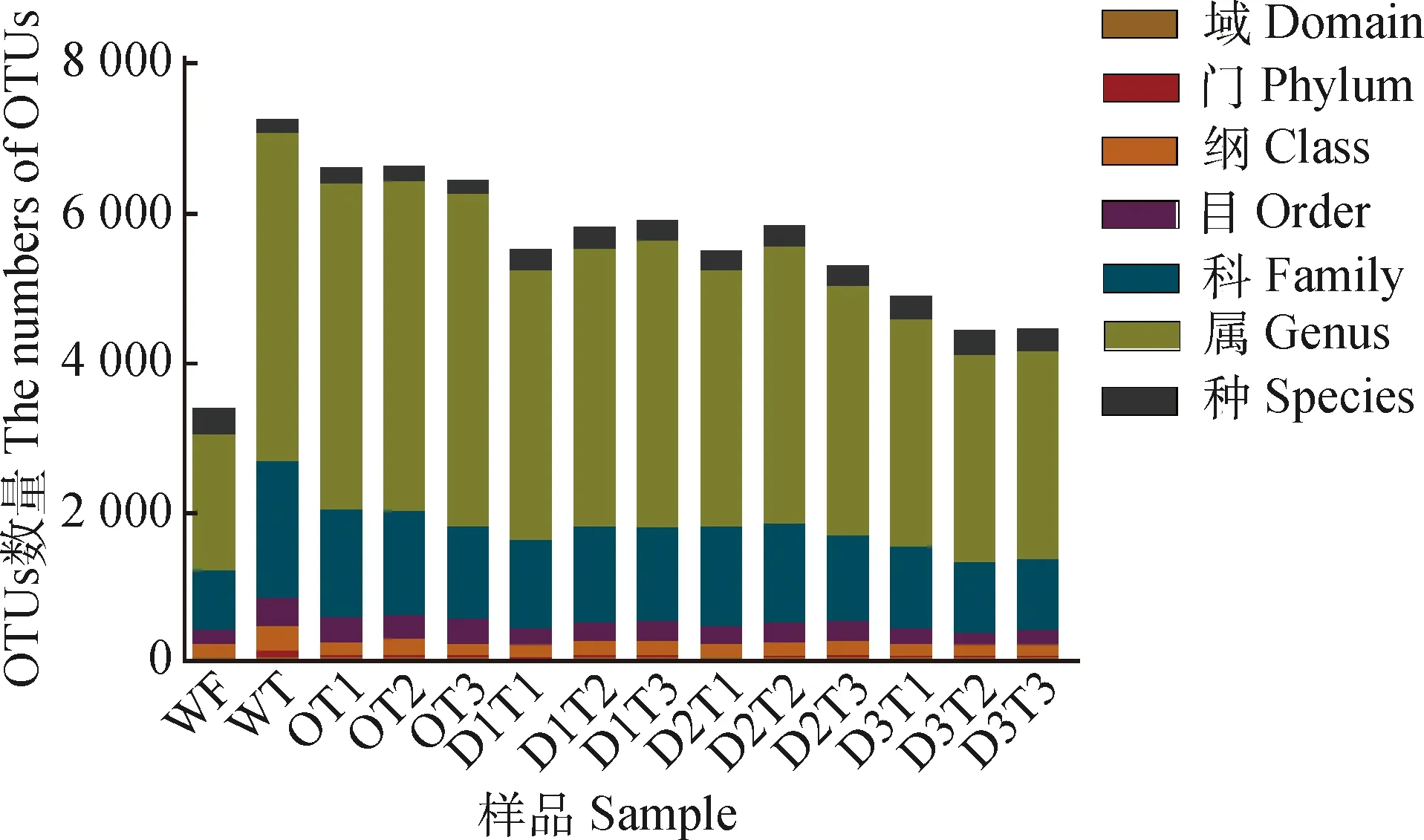
图2 各样品在不同分类水平上的OTU注释数目Fig.2 OTU annotation number at varied classification level of different samples
2.3 物种分类学组成分析
由图3所示,样品中鉴定到的OTU主要分布在以下10个门:放线菌门(Actinobacteria)、变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)、绿弯菌门(Chloroflexi)、酸杆菌门(Acidobacteria)、拟杆菌门(Bacteroidetes)、芽单胞菌门(Gemmatimonadetes)、Patescibacteria、疣微菌门(Verrucomicrobia)、消化螺旋菌门(Nitrospirae)。其中,Actinobacteria最为丰富,在14个样品中相对丰度为55.9%~32.2%;其次为Proteobacteria,相对丰度为36.9%~20.5%。

图3 样品在门水平的群落相对丰度Fig.3 Relative abundance of samples bacterial diversity at phylum level
Actinobacteria和Proteobacteria也是样品WT与WF的优势菌门,Proteobacteria,Firmicutes和Bacteroidetes在WF种的相对丰度分别高于在WT中的丰度。与O组相比,添加牛粪后D1,D2和D3组的Firmicutes和Bacteroidetes相对丰度显著增加(P< 0.05),Proteobacteria在D3组达到显著性增加(P< 0.05);Chloroflexi,Acidobacteria,Gemmatimonadetes,Verrucomicrobia,Nitrospirae丰度呈显著性下降(P< 0.05),Actinobacteria在D3组下降显著(P< 0.05)。Patescibacteria变化不显著(表1)。

表1 不同添加量及取样时间下各样品在门水平的相对丰度Table 1 Relative abundance of bacterial taxa classified to the phylum level at different adding amount and sampling time
随着取样时间的延伸,Firmicutes,Chloroflexi,Gemmatimonadetes,Patescibacteria的相对丰度呈现显著增加(P< 0.05),Bacteroidetes呈现下降趋势。
在属分类水平上,丰度较低的稀有种群占78.03%~84.67%,表明土壤中存在大量待发掘的细菌物种。为了便于分析鉴定到的优势菌群对添加牛粪及取样时间的响应,将相对丰度位于前10位的优势菌属作图。如图4所示,样品WT及WF具有不同的优势菌属,WT的优势菌属有Subgroup_6(酸杆菌)、鞘氨醇单胞菌属(Sphingomonas)、假诺卡氏菌属(Pseudonocardia)、芽球菌属(Blastococcus)、KD4-96(绿弯菌)、67-14(放线菌);WF中具有相对丰度较高的芽孢杆菌属(Bacillus)、马杜拉放线菌属(Actinomadura)、BIrii41(厚壁菌)。

图4 样品在属水平的群落相对丰度Fig.4 Relative abundance of samples bacterial diversity at genus level
Bacillus和Actinomadura的相对丰度均随着牛粪添加比例的增加而升高,且三个水平添加组显著高于O组(P< 0.05);德沃斯氏菌属(Devosia)与BIrii41在三个添加组中的丰度也均高于O组。Subgroup_6,Pseudonocardia,Blastococcus,67-14,KD4-96的相对丰度均随着添加比例的增加呈显著下降,Sphingomonas丰度在添加至D2组时显著低于O组(表3)(P< 0.05)。
从取样时间上看,Sphingomonas和Devosia的相对丰度受取样时间影响显著,随取样时间延长呈显著下降(P< 0.05)。Blastococcus相对丰度在30 d和90 d呈显著性差异(P< 0.05)。Pseudonocardia,Actinomadura和绿弯菌KD4-96的相对丰度在三个取样时间上没有显著差异。Subgroup_6,Bacillus,BIrii41和67-14的相对丰度随取样时间延长呈增加趋势。

表2 不同添加量及取样时间样品在属水平的相对丰度Table 2 Relative abundance of bacterial taxa classified to the genus level at different adding amount and sampling time
2.4 Alpha多样性分析
与O组相比,不同添加比例对Alpha多样性指数Chao1,Observed_species,Shannon,Simpson均产生显著性的影响(P< 0.05)(表3)。O组的四个指数均为最高,随着土壤中牛粪含量的增加,四个指数均呈明显下降趋势,表明细菌群落的丰富度及多样性在下降(P<0.05)。四个指数在D1组和D2组之间差异不显著。

表3 不同添加比例细菌Alpha多样性指数Table 3 Bacterial Alpha diversity indices at different adding amount
2.5 Beta多样性的主坐标轴分析
基于样本间Bray-Curtis距离,PCoA主坐标分析样本群落之间的相似度和差异性。由图5所示,主坐标轴PCo1可解释全部样品细菌群落丰度和多样性方差的22.4%,主坐标轴PCo2可解释细菌群落丰度和多样性方差的的11.1%。不同样本处理组群落组成及多样性之间明显分离,可以分为3大区域:样本D3组(第四象限)主要分布在主坐标PCo1附近,说明D3组苜蓿根际土壤细菌群落结构受PCo1影响明显。第二区域主要分布在PCo2附近(第三象限),即O组在PCo2主轴上分开,说明O组根际土壤细菌群落结构主要受PCo2影响。第三区域为D1组和D2组(第一象限),距离PCo1及PCo2上均较远,D1组内样本间距离也较远,表明D1组的细菌群落结构差异大于D2组。
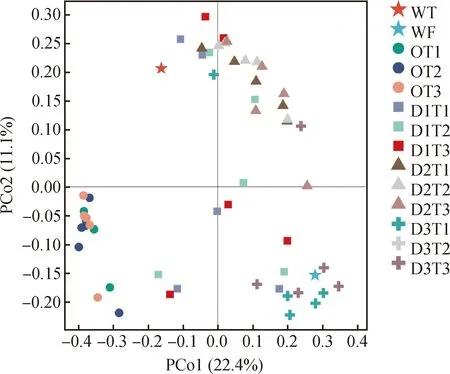
图5 基于Bray-Curtis差异PCoA分析土壤样品细菌群落Fig.5 PCoA analysis with Bray-Curtis dissimilarity of the bacterial communities between samples
2.6 样本间物种差异分析
为研究不同样本间共有及独有的物种情况,使用OTU丰度表制作花瓣图进行群落分析。如图6所示,不同比例牛粪在添加30 d,OT1组具有最多的OTU分布数目,其次为D3T1组,D1T1组为最少,四组一共有OTU 45 004个,共有OTU数目为1 880个,约占4.2%。添加60 d,OT2组具有最多的OTU分布数目,其次为D1T2组,D3T2组为最少,四组一共有47 737个,共有的OTU数目为1 464个,约占3.1%。添加90 d,OT3组具有最多的OTU分布数目,其次为D1T3组,D3T3组为最少,四组一共有OTU 48 321个,共有的OTU数目为1 331个,约占2.7%。从时间看,随着取样时间的延伸,D1组(添加3%)和D2组(添加6%)的OTU数目呈增加趋势,D3组(添加9%)OTU呈下降趋势;四组样本共有的OTU数目比例下降,说明物种的多样性呈增加趋势。
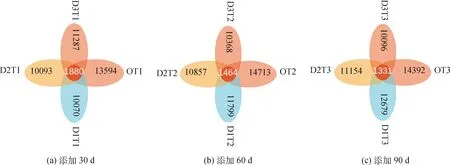
图6 不同取样时间下样本间共有和独有OTU数目的花瓣图Fig.6 The petals figure based on common or unique OTU number among treatments at sampling time
2.7 物种组成差异热图
为了进一步比较样本间的物种组成差异,将平均丰度前20位的属的丰度数据进行聚类分析(图7)。如图7所示,Lysobacter,BIrii41,Devosia,Steroidobacter的丰度在WF与WT及O组间相似,在添加牛粪后丰度增加。Cellvibrio(纤维菌属)、Planifilum(直丝菌属)、Bacillus、Saccharomonospora(糖单胞菌属)丰度在WF,WT及O组差异不明显,在土壤中添加牛粪后,明显高于WF,WT,O组,并随着牛粪含量的增加呈增加趋势,在D3组处理中均达到最高峰,表明混合土壤会促进这些菌属的丰度。Actinoplanes(游动放线菌属),67-14,Blastococcus,KD4-96,Skermanella(红弧菌属),RB41,Subgroup_6,Pseudonocardia(假诺卡氏菌属)的丰度在WT中含量最高,随着牛粪比例的增加明显下降,在WF组丰度最低。Actinomadura和Haloactinopolyspora在WF中高丰度的细菌。

图7 属水平物种组成差异热图Fig.7 Heatmap tree for different treatments cluster analysis at genera level注:标尺颜色深浅表示样本物种丰度变化Note:Scale plate color shades indicate changes in species abundance of samples
与WT相比,O组的Skemanella,Subgroup_6,Pseudonocardia,Sphingomonas丰度增加,Actinoplanes和Pseudomonas丰度下降,其余细菌丰度变化不明显。
3 讨论
施用有机肥是提高作物产量及改善土壤理化性质的主要措施,不同类型肥料及施用时间对土壤微生物群落的影响不同。有研究结果显示,短期施肥对土壤微生物的影响不显著,长期合理施肥可以丰富土壤微生物多样性,恢复土壤中固氮菌群的多样性和群落结构[14,17]。也有研究表明施肥对影响土壤细菌群落丰度、土壤微生物多样性的影响具有一定的局限性[18-19]。本研究结果表明腐熟牛粪在短期内降低苜蓿根际土壤细菌Alpha多样性。这与赵凤艳等研究结果相似,牛粪降低设施番茄根际细菌多样性和丰富度[20]。Zhang等[21]在茶园施用1.5%牛粪和尿素后,Alpha多样性指数均低于未施肥,且在取样时间上土壤细菌的多样性变化趋势相似:春季至夏季显著增加,夏季至秋季显著减少。也有研究发现细菌丰富度指数Chao和ACE两个指数在添加固体牛粪和猪粪后下降,在取样季节之间差异显著,在不同肥料间差异不显著[22]。类似研究结果显示季节对细菌群落的影响大于肥料的营养处理[23]。本研究的多样性指数和丰富度指数均呈现O> D1> D2> D3,3%的牛粪添加量可使Alpha多样性显著下降,土壤细菌的群落结构发生显著变化。一方面是由于盆栽种植苜蓿,土壤细菌菌群的丰度及多样性变化会更加显现;另一方面可能是本试验施用了相对较高的肥料添加比例导致。
Actinobacteria是所有测试样品中的优势菌群,在WT和WF中相对丰度分别达56%和42%,并且随着牛粪添加比例的增加而呈下降,添加至9%时与O组差异达到显著。随着取样时间的延长也表现下降趋势。Actinobacteria功能多样,有助于有机物的分解,研究表明Actinobacteria在未受到干扰的土壤中易富集[24]。Proteobacteria是测试样品中的另一优势菌群,在WT中相对丰度最低。Proteobacteria能够适应营养丰富的环境,Acidobacteria通常被认为喜欢资源有限的寡养环境[25-27]。前人研究发现Proteobacteria与Acidobacteria的比例可以反映土壤营养状况:二者比例越高意味着更多的有机质投入[28]。本研究也证实了这结论,随着牛粪添加比例的增加,Proteobacteria丰度增加,Acidobacteria丰度降低,二者比例呈增加趋势。
Bacteroidetes是一类厌氧菌,菌门成员具有降解复杂有机化合物如纤维素的功能[23]。本研究结果表明土壤中添加牛粪显著增加Bacteroidetes丰度。Firmicutes也是一类厌氧菌,高碳利用率可能导致其丰度增加。本研究发现随取样时间延长,Firmicutes丰度表现增加趋势,30 d与90 d达到显著性差异,推测在苜蓿的一个生长周期,碳利用率在持续增加。Gemmatimonadetes可以对土壤中碳和氮含量进行微调,平衡作物代谢需求,适应不断变化的环境,对土氮素循环中反硝化作用具有重要意义[29]。添加牛粪后Gemmatimonadetes相对丰度显著下降,但随着时间延长呈显著增加,受到作物生长阶段影响显著。Chloroflexi和Patescibacteria的相对丰度随作物生长呈现显著增加,这一结果与刘静平等研究结果相似[30]。但添加牛粪会显著降低Chloroflexi丰度。Tian等[31]发现其相对丰度与有机肥施用量呈负相关关系。Chloroflexi作为重要的有机物分解者,和放线菌一样对有机肥施用非常敏感[32],施用有机肥反而会抑制Chloroflexi和Actinobacteria的生长和繁殖,降低二者相对丰度。Nitrospirae是共营养细菌,适合高pH值环境条件生存,施肥降低其丰度[33-34]。本研究结果表明添加牛粪会降低其丰度。
在属水平上,WT及WF具有不同的优势菌属。添加牛粪的土壤可显著增加有益菌属芽孢杆菌属(Bacillus)、马杜拉放线菌属(Actinomadura)、BIrii41、德沃斯氏菌属(Devosia),降低Subgroup_6、Pseudonocardia、芽球菌属(Blastococcus)、67-14、KD4-96(绿弯菌)的丰度。变形菌门根瘤菌目Devosia丰度在WT及WF样品中均很低,但在添加牛粪土壤中,该菌属的丰度显著增加,达45倍以上,但也随着苜蓿生长显著下降。Devosia与水生豆科植物Neptunianatans形成独特的固氮共生体,有推测Devosia可能有助于速效氮含量的增加[35]。BIrii41显示与Devosia相同变化,在添加牛粪土壤中,该菌属的丰度显著增加,并且随苜蓿生长时期延长呈增加趋势。厚壁菌门中与有机化合物分解和植物抗病性相关的Bacillus和对温度适应的多样性Planifilum,放线菌门中能降解半纤维素和蛋白质的Saccharomonospora,变形菌门中与植物的抗病性相关的Lysobacter、降解纤维素的Cellvibrio和具有反硝化能力的Steroidobacter[36],这些菌群的丰度在WF,WT,O组差异不显著,在土壤中添加牛粪后,显著高于WF,WT,O组,并随着牛粪含量升高而增加。虽然添加牛粪降低Sphingomonas的丰度,但该菌属的丰度变化并没有随添加比例呈现规律性变化,反而随着取样时间延伸呈现出显著性的降低。
4 结论
添加腐熟牛粪降低盆栽苜蓿土壤细菌Alpha多样性。Actinobacteria和Proteobacteria是土壤、腐熟牛粪及混合土壤样品的优势菌。添加腐熟牛粪增加土壤Firmicutes,Bacteroidetes和Proteobacteria丰度;显著提高Lysobacter,Bacillus,Saccharomonospora,Planifilum,Cellvibrio,Steroidobacter,Devosia的丰度。不同添加比例均降低土壤细菌OTU数量,但添加比例至6%牛粪的土壤细菌OTU数目随着苜蓿生长持续增加,高添加比例(添加9%)的细菌OTU数目持续降低。通过不同添加比例的腐熟牛粪对苜蓿土壤细菌结构及多样性的影响分析,推荐腐熟牛粪添加量低于6%的施肥方式为佳。
