微波辐射无皂乳液聚合法制备PS微球
2022-03-21贾慧灵梁梦颢吴锦绣谭心胡金豹
贾慧灵,梁梦颢,吴锦绣,谭心,胡金豹
(1.内蒙古科技大学机械工程学院,内蒙古包头 014010; 2.轻稀土资源绿色提取与高效利用教育部重点实验室,内蒙古包头 014010;3.内蒙古自治区稀土湿法冶金与轻稀土重点实验室,内蒙古包头 014010; 4.内蒙古科技大学材料与冶金学院,内蒙古包头 014010)
聚苯乙烯(PS)微球具有吸附性强、凝集作用大、比表面积大及表面反应能力强等优点[1],在生物工程、光学材料、化工等领域已有广泛的应用[2–4]。PS微球常用的制备方法包括乳液聚合法、分散聚合法、无皂乳液聚合法、种子聚合和悬浮聚合等方法[5–6]。其中,无皂乳液聚合法在制备单分散性良好的聚合物微球方面具有独特的优势[7–8]。无皂乳液聚合是指在聚合反应过程中不加入乳化剂或加入浓度低于临界胶束浓度的微量乳化剂的乳液聚合过程,该工艺具有生产成本低、合成过程简单、后处理简洁等优点,所制备的微球表面洁净、粒径均匀[9–10]。
洪秀秀[11]采用无皂乳液聚合法,以水浴加热的方式制备出分散性良好的PS微球,并指出微球粒径随着单体用量及反应体系离子强度的增大而增大,随着引发剂浓度的增加而减小,且随聚合温度升高而先增大后减小。李玉等[12]通过加入微量乳化剂或β-环糊精对无皂乳液聚合法进行了改进,制备出粒径在300 nm左右的单分散PS微球,并指出β-环糊精的加入可大幅缩短反应时间。袁媛[13]将可逆加成-断裂链转移自由基和无皂乳液聚合法相结合,采用油浴加热方式制备出表面带有“活性”位点的PS微球,研究了反应时间、温度及过硫酸钾(KPS)与二乙基二硫代氨基甲酸苄酯(BDC)投料比等因素对PS微球的影响。然而,无论是水浴或是油浴加热均是通过热传导的方式使得反应溶液升温,这会导致反应溶液存在受热不均的情况,且传统加热方式下沸腾法无皂乳液制备的PS微球表面凹凸不平、球形度极差[14]。微波辐射反应具有加热速率快、转化率高、能耗低等优点[15]。近年来,采用微波辐射加热的方式已开始应用于聚合研究领域中,如微波辐射乳液聚合、微波辐射分散聚合[15–16],易昌凤等[17]、邓字巍等[18]采用分散聚合法以微波辐射加热的方式制备了粒径在200~500 nm的单分散PS微球,并指出微波辐射加热得到的微球粒径及单分散性要优于常规加热制备的微球。
笔者采用无皂乳液聚合法,旨在改善采用沸腾无皂乳液聚合法制备PS微球时,微球表面形貌不光滑且球形度极差的情况。以微波加热方式代替传统油浴或水浴加热方式,研究了引发剂浓度、交联剂与稳定剂用量及微波功率大小对PS微球粒径及形貌的影响,分析了不同工艺条件下PS微球表面Zeta电位,确定了最优工艺条件下制得的PS微球的力学性能。为制备出表面形貌良好、单分散性分布的PS微球提供了理论依据。
1 实验部分
1.1 原材料
苯乙烯:分析纯,天津市大茂化学试剂厂;
氢氧化钠(NaOH):分析纯,天津市化学试剂三厂;
二乙烯苯(C10H10)、α-甲基丙烯酸(C4H6O2)、KPS:分析纯,上海麦克林生化科技有限公司;
去离子水(H2O):自制。
1.2 仪器及设备
微波仪:MEBT-Ⅲ型,北京纽比特科技有限公司;
傅里叶变换红外光谱(FTIR)仪:Nicolet iS50型,美国赛默飞世尔科技有限公司;
场发射扫描电子显微镜(FESEM):SIGMA500型,德国卡尔蔡司公司;
激光粒度仪:LS230型,美国贝克曼库尔特有限公司;
Zeta电位分析仪:Zetasizer Nano ZS ZEN3600型,英国马尔文仪器有限公司;
原子力显微镜:Cyper ES 型,美国牛津仪器公司。
1.3 PS微球的制备
量取90 mL去离子水、20 mL苯乙烯、2 mL二乙烯苯、1 mL α-甲基丙烯酸依次加入到500 mL带有冷凝管及磁力搅拌装置的三口烧瓶中,超声分散15 min后置于微波仪中,在温度100℃、磁力搅拌转速为970 r/min的情况下加热至沸腾,15 min后加入0.2 g引发剂KPS (需先溶于10 mL去离子水中),反应2 h后得到PS微球乳胶液。
1.4 样品表征
FTIR分析:分辨率为4 cm–1,扫描范围为4 000~400 cm–1;
SEM表征:将样品涂在导电胶上做喷金处理,对样品表面形貌、粒径尺寸及分散性进行观察;
粒径测试:将样品超声分散在无水乙醇中,采用激光粒度仪按照GOST R 8.777–2011测定样品粒径,计算粒径分布宽度(R)=[(D90–D10)/D50],D50表示样品的累计粒度分布达到50%时所对应的粒径,被称为中心粒径,用来表示粉体的平均粒度,D90则表示样品的累计粒度分布达到90%时所对应的粒径,用来表示粉体粗端的粒度,D10表示样品的累计粒度分布达到10%时所对应的粒径;
微球表面电势用电位分析仪测定;
力-位移曲线测定:采用原子力显微镜,用NanoScope Analysis软件分析力-位移曲线的数据;
压缩弹性模量按照GB/T 1041–2008测试,根据Hertz接触模型计算。
2 结果与讨论
2.1 PS微球的FTIR分析
PS微球的FTIR谱图如图1所示。由图1可知,1 603.1,1 493.8 cm–1和1 451.5 cm–1为苯环上C=C的伸缩振动峰,3 084.2,3 062.7 cm–1和3 025.3 cm–1为苯环上不饱和C—H的伸缩振动峰,2 927.1,2 855.2 cm–1分别为烷烃类C—H的反称与对称伸缩振动峰,757.2 cm–1和698.9 cm–1为单取代苯环C—H面外弯曲振动峰,而1 068.4 cm–1和1 026.1 cm–1为单取代苯环C—H的面内弯曲振动峰,542.4 cm–1为乙烯化合物的C=C的扭曲振动峰,这些特征峰的出现说明苯乙烯发生聚合反应,所得样品为PS。由图1还可以看出,3 442.7 cm–1为O—H键的伸缩振动峰,说明聚合物表面存在羟基,1 701.3 cm–1处的小峰为C=O的伸缩振动峰,说明所制备的样品中含有羧基,这些羟基与羧基的产生是由于KPS为水溶性无机过氧化类引发剂,受热分解产物为—SO4–,既是离子又是自由基,在水中这些初级自由基可与水分子反应形成—OH,而羟基进一步被氧化为弱酸性的羧基;羧基也有可能来自稳定剂α-甲基丙烯酸中的羧基官能团。
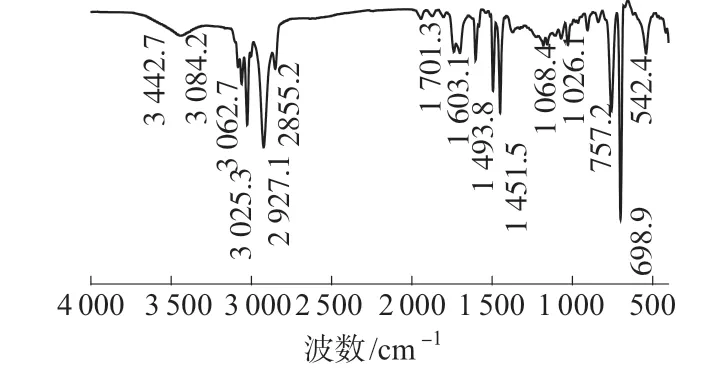
图1 PS微球的FTIR谱图
2.2 不同工艺对PS微球粒径及形貌的影响
(1)引发剂浓度。
当交联剂二乙烯苯与稳定剂α-甲基丙烯酸体积比为1∶1,微波功率为300 W时,分别研究引发剂KPS浓 度为3.03×10–3,6.06×10–3,12.13×10–3,18.19×10–3mol/L时的PS微球形貌和粒径。
图2为不同引发剂浓度下所制备的PS微球的SEM照片。由图2a和图2b可看出,当KPS浓度(3.03×10–3mol/L)较低时,PS微球的球形度较差且粒径不均匀,随着KPS浓度的增加,PS微球的团聚现象有明显的改善且球形度变好、表面光滑且粒径均匀。由图2c可看出,进一步增加KPS浓度,PS微球粒径变得不均匀,且球形度变得较差,表面也变得不光滑。KPS浓度增加到18.19×10–3mol/L时,发现大量的PS微球显著地团聚在一起(图2d)。

图2 不同KPS浓度下PS微球的SEM照片
图3为不同KPS浓度下所制备的PS微球的粒径分布图。由图3a可看出,当KPS浓度为3.03×10–3mol/L时,PS微球的粒径分布图出现了双峰,出现双峰是因为KPS浓度较低,自由基—SO4–产生的速率较慢,与苯乙烯单体结合的几率较低,成核速度较慢,生成一些粒径较大且形状不规则的颗粒,则导致制备的PS微球粒径不均匀(图2a)。当KPS浓度为6.06×10–3mol/L时,PS微球的粒径分布图呈现单峰,说明KPS在该浓度下所制备的PS微球粒径单分散性较好(图2b)。当KPS浓度增加到12.13×10–3mol/L时,其粒径分布图又出现双峰(图3c),是因为随着KPS浓度的增加,自由基—SO4–的产生速率加快,与苯乙烯单体的结合几率增大,聚合反应速度加快,而苯乙烯单体总含量不变,这就使得成核粒子中所包含的单体量减少,生成一些粒径较小的颗粒(图2c)。当KPS浓度继续增加到18.19×10–3mol/L时,样品粒径分布更不均匀,一方面是由于小粒径的生成,另一方面PS微球还出现了明显的团聚现象(图2d),这导致其粒径分布曲线中产生多个杂峰。
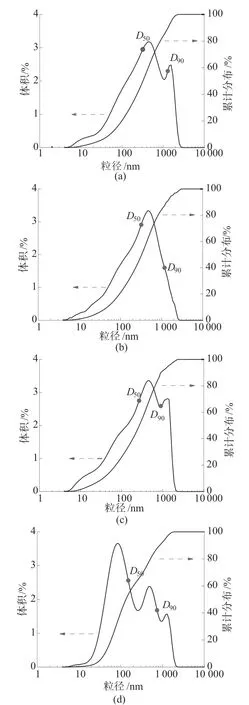
图3 不同KPS浓度下PS微球的粒径分布图
表1为不同KPS浓度下所制备的PS微球的中心粒径D50和R数据。由表1可以看出,PS微球的中心粒径随着KPS浓度的增加呈现减小的趋势。这是由于KPS浓度增加,成核粒子数增多,且PS表面—SO4–增多,小粒径的PS微球能够稳定存在。而PS微球的粒径分布宽度则随着KPS浓度的增加呈现先减小后增大的趋势,在6.06×10–3mol/L处粒径分布宽度取得最小值。相应的PS微球的中心粒径为280 nm,这比SEM检测的PS平均粒径240 nm(图2b)偏大。

表1 不同KPS浓度下PS微球的中心粒径及粒径分布宽度
(2)交联剂与稳定剂用量。
当KPS浓度为6.06×10–3mol/L,微波功率为300 W,分别研究交联剂二乙烯苯与稳定剂α-甲基丙烯酸体积比为0.7∶1,1.5∶1,2∶1,3∶1时的PS微球形貌和粒径。
不同交联剂与稳定剂用量下所制备的PS微球的SEM照片如图4所示。由图4a和图4b可看出,随着交联剂与稳定剂体积比从0.7∶1增加到1.5∶1,PS微球团聚现象有了明显的改善,微球表面也变得相对光滑,微球的粒径也有明显减小的趋势。当交联剂与稳定剂体积比变为2∶1时,PS微球的粒径变得均匀,且球形度较高,微球表面变得更光滑,微球间的团聚现象得到改善(图4c)。交联剂用量继续增加,由图4d可以看出,微球的形貌开始变差,出现了团聚现象,且微球表面也不再光滑。

图4 不同交联剂与稳定剂用量下PS微球的SEM照片
不同交联剂与稳定剂用量下PS微球的粒径分布如图5所示。当交联剂与稳定剂体积比为2∶1时,PS微球的粒径分布出现一个单峰,而在其余交联剂与稳定剂体积比下粒径分布均出现了多峰。出现该情况的原因是,当交联剂与稳定剂体积比为0.7∶1时,聚合物交联度较低,捕获单体或自由基的效率较高,PS微球生长形貌较差、粒径分布不均匀(图4a)。随着交联剂用量的增加,聚合物增长减慢,PS微球粒径变小且粒径分布变得均匀(图4c)。当交联剂与稳定剂体积比增加到3∶1时,聚合物的交联度过高,会阻碍与自由基和聚合物链的结合效率,导致二次成核,使得粒径分布图出现杂峰(图5d)。
表2为不同交联剂与稳定剂体积比下PS微球的中心粒径和R数据。由表2可看出,PS微球的中心粒径和粒径分布宽度均呈现先减小后增大的趋势。当交联剂与稳定剂体积比为2∶1时,中心粒径达到最低为240 nm。粒径分布宽度值也达到最低,这与粒径分布图5c的单峰分布相一致。
经由我院伦理委员会批准从本院2010年1月至2017年12月接受的终末期糖尿病肾病血液透析患者中,抽取52名,随机将其分为对照组与观察组,均26例。对照组中,男14例,女12例,年龄48~75岁,平均年龄(61.5±13.5)岁,病程2~5年,平均病程(3.5±1.5)年。观察组中,男14例,女12例,年龄47~75岁,平均年龄(61±14)岁,病程2.5~4.5年,平均病程(3.5±1.5)年;两组一般资料比较结果p>0.05,可作比较。
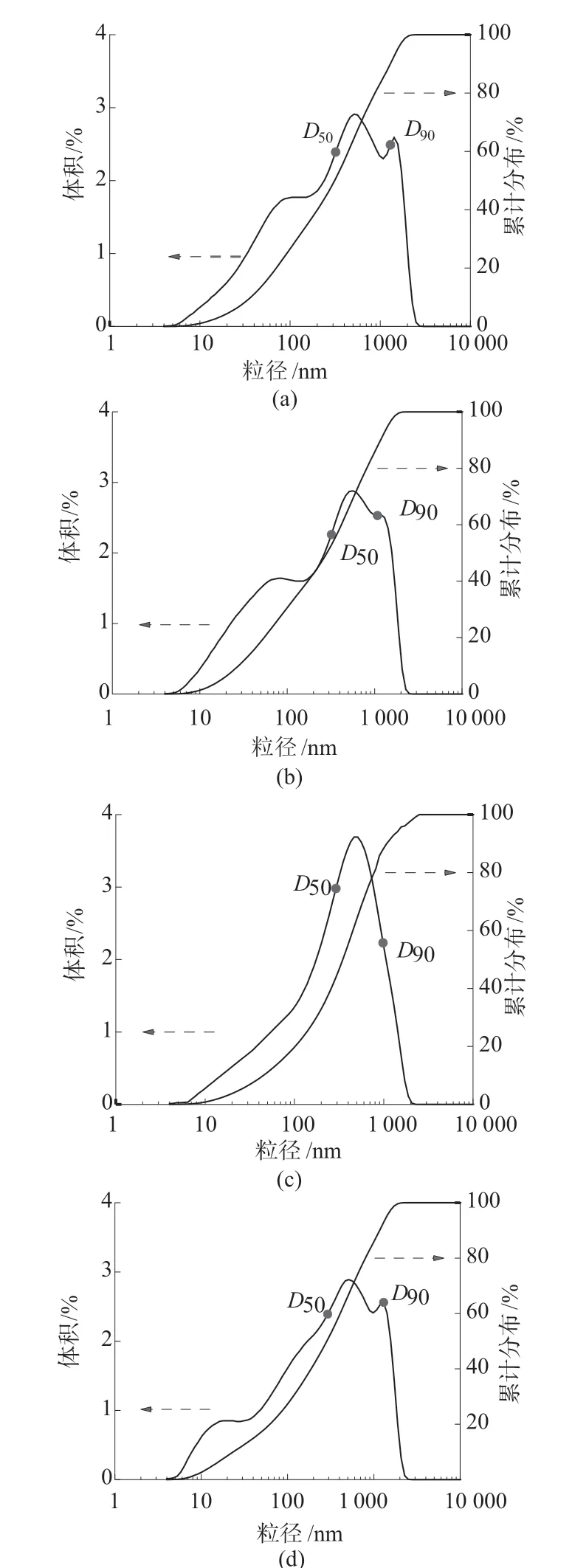
图5 不同交联剂与稳定剂用量下PS微球的粒径分布图
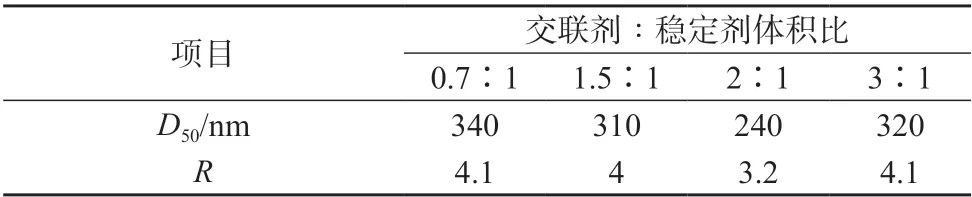
表2 不同交联剂与稳定剂用量下PS微球的中心粒径及R
(3)微波功率。
当引发剂浓度为6.06×10–3mol/L,交联剂与稳定剂体积比为2∶1时,分别研究微波功率为250,300,350 W时的PS微球形貌和粒径。
图6为不同微波功率下所制备的PS微球的SEM图。由图6a可以看出,在功率为250 W下所制备的PS微球表面不光滑、球形度较差且粒径分布不均匀。由图4c可以看出,微波功率达到300 W时,PS微球表面变得光滑、球形度较高且粒径均匀性良好。微波功率增大到350 W时,虽然PS微球表面依旧光滑,但其球形度变差且粒径变得不均匀。综上所述,采用合适的微波辐射加热方式所制备的PS微球的表面形貌及球形度均优于文献[14]中采用油浴加热方式所制备的PS微球。

图6 不同微波功率下PS微球的SEM图
图7为不同功率下所制备的PS微球的粒径分布图。从图7可以看出,当功率为250 W和350 W时,PS微球的粒径分布度均出现了杂峰,表示在这两种情况下所制备的PS微球的粒径存在分布不均匀的情况。这是由于功率较低(250 W)时,反应体系升温较缓慢,使得聚合过程缓慢进行,导致PS微球粒径生长不均匀且形貌较差(图6a)。而当功率较高(350 W)时,反应体系快速达到聚合反应温度,聚合过程变得剧烈,会产生一些较小的颗粒且导致颗粒球形度变差(图6b)。当功率为300 W时,其粒径分布曲线只有一个单峰(图5c),说明该功率下所制备的PS微球粒径分布较均匀。
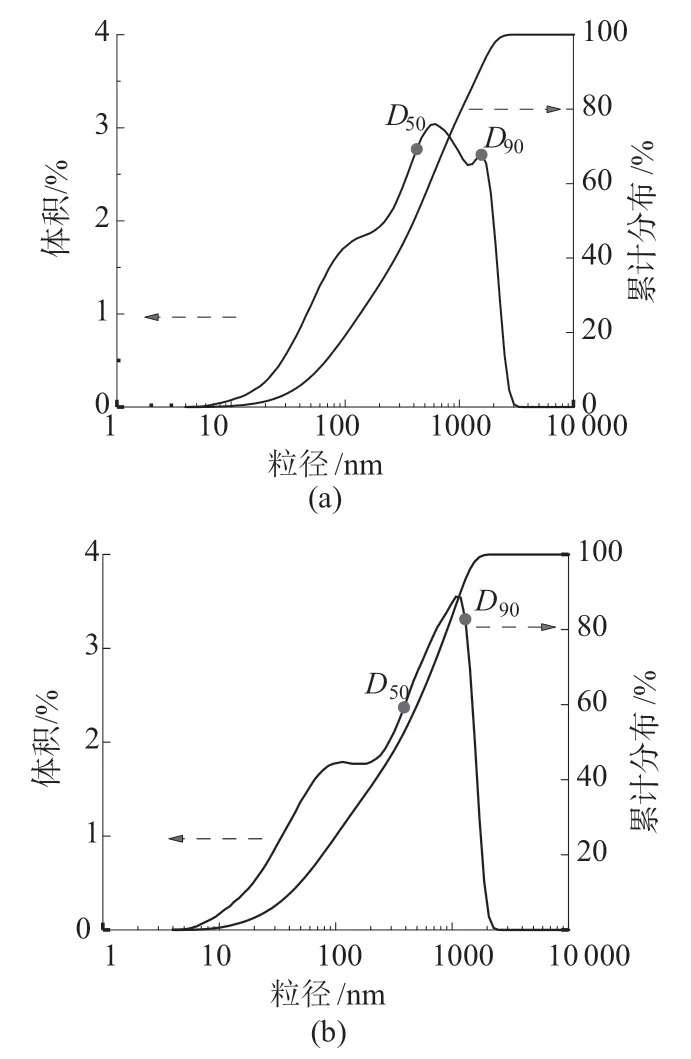
图7 不同功率下PS微球的粒径分布图
不同微波功率下PS微球的中心粒径和粒径分布宽度的数据列于表3。由表3可知,当微波功率为300 W时,中心粒径达到最小值为240 nm。粒径分布宽值达到最小。综合上述分析可得出,微波功率为300 W时所制备的PS微球表面形貌最好、粒径分布最均匀。

表3 不同微波功率下PS微球的中心粒径及R
2.3 PS微球电位分析
表4为不同工艺下制备的PS微球的Zeta电位数据。由表4数据表明,PS微球表面均是显电负性,这是由于引发剂KPS热分解产生了自由基—SO4_聚合反应过程中PS微球表面引入了—SO4–基团。当引发剂浓度为6.06×10–3mol/L时,PS微球表面Zeta电位的绝对值达到最大,表明该聚合物的稳定性较好,从图2b也可看出,PS微球具有良好的分散性。同样,当交联剂与稳定剂的体积比为2∶1、微波功率为300 W时,PS微球表面Zeta电位值为–37.4 mV,其绝对值也保持最大,这从另一方面解释了图5c中PS微球粒径分布呈现单分散性的原因。

表4 不同工艺下所制备的PS微球的电位
2.4 PS微球的压缩弹性模量分析
对PS微球样品进行力-位移曲线测定时,微悬臂的探针接近、压入和离开样品表面,微悬臂和样品之间会在载荷力作用下产生相对位移,可依据Hooke定律由公式(1)计算出探针与被测样品之间的作用力(F),从而得到力曲线。图8为所测得的最优工艺条件下制备的PS微球的力-位移曲线。

图8 PS微球的力-位移曲线图

式中:Kc——探针的弹性系数;
d——微悬臂的偏移量;
d0——微悬臂的初始偏移量。
测试选用圆锥形针尖,根据Hertz接触理论,载荷F与压痕深度δ之间的关系如式(2)所示。

式中:α——针尖的圆锥半角,测试选用ACTG型探针针尖的圆锥半角为17°;
E*——等效弹性模量,其值可通过公式(3)计算。

式中:E1和E2——探针和被测样品的压 缩 弹 性 模量;
ν1和ν2——探针和被测样品的泊松比。
针尖压入样品表面的压痕深度δ由公式(4)计算。
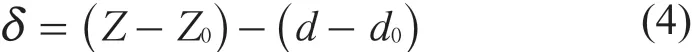
式中:Z——样品的高度;
Z0——样品的初始高度偏移量。
利用AFM软件对力曲线进行数据处理,得到不同位置的d–d0及Z–Z0的值,带入公式(4)中,计算出不同位置的压痕δ值,作出PS微球的载荷-压痕曲线,如图9所示。借助公式(2),由载荷及压痕得出PS微球的等效弹性模量E*,再通过公式(3)得到最优工艺条件下制备的PS微球的压缩弹性模量E为2.75 GPa,这与陈杨等[19]对亚微米级PS微球的压缩弹性模量分析结果(2.01~2.90 GPa)基本一致。

图9 PS微球的载荷-压痕曲线
3 结论
采用无皂乳液聚合法,通过调节引发剂浓度为6.06×10–3mol/L、交联剂与稳定剂体积比为2∶1及微波功率为300 W制备出球形度较高、表面光滑、粒径分布均匀且单分散性高、表面带负电荷的PS球,微球的平均粒径约为240 nm。研究结果如下:
(1)随着引发剂KPS浓度的增加,PS微球粒径呈现减小的趋势。当引发剂KPS浓度过高时,小粒径的PS微球出现团聚现象,使得微球粒径分布变宽。
(2)随着交联剂与稳定剂体积比的增加,PS微球粒径呈现先减小后增大的趋势,在体积比为2∶1时所制备的PS微球表面形貌及分散性良好,当交联剂用量继续增加,交联度过大会导致聚合过程中二次成核,微球的粒径分布宽度增大,表面形貌变差。
(3)较高或较低的微波功率都会导致PS微球的粒径分布不均匀,过低的微波功率还会导致微球表面不光滑。
(4) PS微球的Zeta电位值均为负值,表明所制备的微球表面带有负电荷。最优工艺下制备的PS微球的Zeta电位值为–37.4 mV,该分散体系具有良好的稳定性,未出现团聚现象。
(5)采用AFM测定PS微球力曲线,根据Hertz接触理论计算得到最优工艺下制备的PS微球的压缩弹性模量为2.75 GPa。
