医药洁净厂房空调系统确认和环境监测取样点选取探讨
2022-03-19任杏珠
任杏珠
(中国电子系统工程第四建设有限公司,河北 石家庄 050000)
0 引 言
医药工业洁净厂房和空调系统主要为药品的生产环境提供洁净要求,从而保证所生产的药品安全、有效和质量可控,满足患者需求。
中国GMP 附录1,无菌药品第三章第九条明确提出,洁净区的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准;第十条要求应对洁净区的悬浮粒子进行动态监测;第十一条要求应当对微生物进行动态监测。
从以上要求可以看出,洁净厂房和空调系统设计后需进行“静态和动态”确认,而日常需进行动态环境监测。但对于确认和日常监测期间悬浮粒子和微生物采样点数量和位置的选择一直是大家讨论的话题。本文将针对该问题,结合国内外不同法规和指南建议进行重点对比阐述。
来自ISO14644-1 2015 版的静态定义:洁净室或洁净区已经建成,生产设备已经安装好,并以指定的方式运行,但没有人员在场。
来自ISO14644-1 2015 版的动态定义:洁净室或洁净区按照规定的方式和既定的条件运行,有设备运行和规定数目的人员在场的状态。
1 静态原则
确认过程中的取样点和位置
1.1 静态尘埃粒子
ISO14644-1 ∶2015(E) 和GB/T 16292 2010《医药工业洁净室(区) 悬浮粒子的测试方法》中分别给出了2 种不同的确定采样点数量和位置的方法。
1.1.1 ISO14644-1:2015(E)
(1) 根据洁净区(室) 面积确定最少采样点数量。
与洁净区面积相关的采样点数量见表1。

表1 与洁净区面积相关的采样点数量Table 1 Sampling locations related to cleanroom area
由表1 可以看出,当洁净室或洁净区面积>1 000 m2的时候,取样点最小数量计算式如下:

式中:NL为待评估的取样点最小数量,上舍入临近整数;A 为洁净室的面积,m2。
最少采样点数量即95%置信区间来保证至少洁净区或洁净室90%面积不超过分类限度。
(2) 采样点的位置布点原则。
根据以上最少采样点数量,将洁净区(室) 分为NL等份,从每个等份中选择采样点作为该等份中的特征代表。在对房间进行等分时,需兼顾房间的布局。在每个等份中,将计数器的探头放在工作平面或者其它指定点。
1.1.2 《医药工业洁净室(区) 悬浮粒子的检测方法》GB/T 16292-2010
(1) 根据洁净区(室) 面积确定最少采样点数量,计算式如下。

式中:NL为最少采样点数(四舍五入为整数);A为洁净室或洁净区的面积,m2。
按照以上方法计算的采样点数与ISO14644-1进行对比,2 种不同的标准计算后采样点数量对比见表2。

表2 2 种不同的标准计算后采样点数量对比Table 2 Comparison of the number of sampling points after two different standard calculations
国标方法计算的采样点数,从洁净室(区) 面积>8 m2开始,均<ISO14644-1∶2015(E) 中的要求。
ISO14644-1∶1999(E) 中关于取样点的相关描述如下:
B4 取样
B4.1 采样点的建立
B4.1.1 根据式(2) 计算最小采样点数量,计算式如式(2)
B4.1.2 确保取样位置均匀的分布在整个洁净室(或洁净区内),位于工作活动面的高度。
如果客户指定额外的取样点,其位置和数量也应指定。
《医药工业洁净室(区) 悬浮粒子的检测方法》GB/T 16292-2010 中关于悬浮粒子取样的原则完全参考了ISO14644-1:1999(E) 中的要求。
(2) 采样点一般在离地面0.8 m 高度的水平面上均匀布置;采样点>5 点时,也可以在离地面0.8~1.5 m 高度的区域内分层布置。同时给出了一个具体的布点方式,可参照进行布点。布点的总体原则上基本一致,均为平均分布。但细分位置描述不如ISO14644-1∶2015(E) 中更具参考意义。采样点布局示意图如图1 所示。

图1 采样点布局示意Fig.1 Sampling point layout diagram
1.2 静态沉降菌最少采样点数和采样位置
《医药工业洁净室(区) 沉降菌的测试方法》GB/T 16294-2010 5.4.1.1 中规定最少采样点数和采样位置均可参考GB/T 16292-2010,同时增加了在满足最少采样点数的同时,还应满足最少培养皿数的要求。最少培养皿数列表见表3。

表3 最少培养皿数列表Table 3 Minimum number of petri dishes list
1.3 静态浮游菌最少采样点数和采样位置
《医药工业洁净室(区) 浮游菌的测试方法》GB/T 16293-2010 5.4.1.1、5.4.1.2 中规定最少采样点数和采样位置均可参考GB/T 16294-2010。
按照ISO 14644-1 2015(E)中悬浮粒子的计算方法确定的最少采样点数,至少保证了95%置信区间来保证至少洁净区或洁净室90%面积不超过分类限度,数量合理,方法更优于国标的计算方式。
通过国标中对沉降菌、浮游菌的取样点和取样位置,参考悬浮粒子。对于沉降菌和浮游菌采样点数、位置也可参考ISO 14644-1 2015(E)中对于悬浮粒子的要求。
2 动态原则
动态测试期间,需模拟正常生产状态最差条件下的人员和可能的动作。
2.1 尘埃粒子及沉降菌和浮游菌取样点数和位置
参考静态测试时洁净区(室) 面积确定最少采样点数量;根据物料暴露区,人员干预区,人员更衣区,房间的特殊布局位置,选择增加额外的取样点,他们的数量和位置应该被批准和说明。此外无菌操作间和背景环境(A 级/B 级) 的取样位置应该考虑所有关键操作区,如位于灌装点的胶塞振动盘。关键工序取样点应基于书面的风险评估,以及对该区域进行的工艺和操作的知识。
2.2 表面微生物
2.2.1 采样点数对于同一洁净区,每个相同的取样物体在其不同的地方至少采2 个样。
2.2.2 取样点布局
洁净室的大小、设备管路等的复杂程度;生产活动的重要性、易受污染的部位。具体位置应评估确定,考虑包含门、门把手、地板、墙壁(不易被清洁/消毒的部位,至少2 点)、公用介质的管路(不易被清洁/消毒的部位)、生产设备的关键部位和五指手套等。
3 监测标准
3.1 悬浮粒子
3.1.1 中国GMP2010 版
中国GMP2010 版附录1 中对于确认过程中的粒子要求,其中中国药典2020 年版,也同样执行以上规定。中国药典中悬浮粒子的标准见表4。

表4 中国药典中悬浮粒子的标准Table 4 Standards for suspended particles in Chinese pharmacopoeia
A 级洁净区空气悬浮粒子的级别为ISO 4.8,以≥5.0μm 的悬浮粒子为限度标准(所以每个采样点的采样量不得少于1 m3)。
B 级区(静态) 的空气悬浮粒子的级别为ISO 5,同时包括表中2 种粒径的悬浮粒子。测试方法可参照ISO 14644-1。
3.1.2 欧盟GMP2020 年
欧盟GMP2020 年附录1 草案,无菌药品中则提出了新的标准。
欧盟GMP 附录1 中悬浮粒子标准见表5。

表5 欧盟GMP 附录1 中悬浮粒子标准Table 5 Standard for suspended particles in Appendix 1 of EU GMP
洁净室分级,对于静态A 级和B 级区域,分级测试应包括对≥0.5 μm 的颗粒进行测量;但可考虑使用更大的第二个粒径,如符合ISO 14644 的1 μm。该测量应在静态和运行状态下进行。
3.1.3 ISO 14644-1
ISO 14644-1 中对洁净室分级确认时的要求,通过粒子浓度进行的空气洁净度ISO 分级见表6。
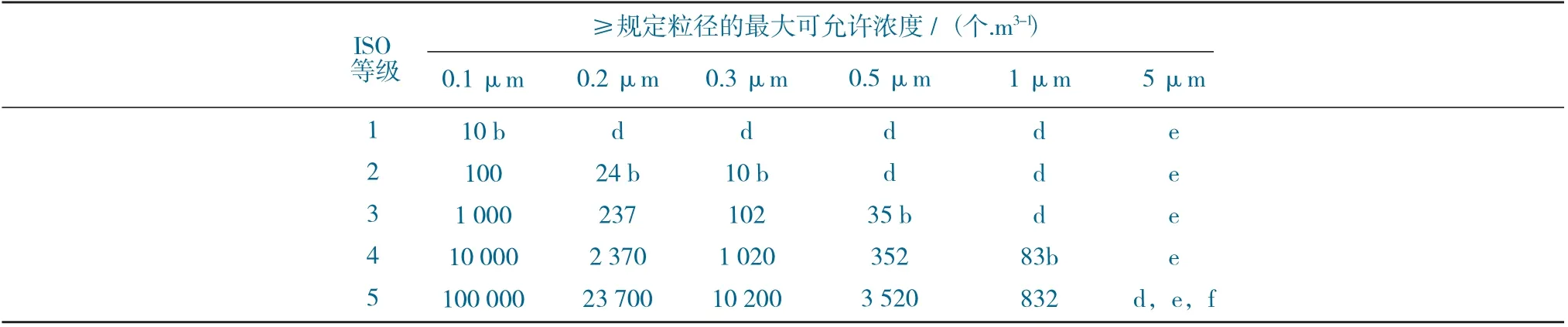
表6 通过粒子浓度进行的空气洁净度ISO 分级Table 6 ISO Classes of air cleanliness by particle concentration

续表
由表6 可以看出以下3 点。
(1) 低浓度粒子,存在取样和统计的局限性,会造成分级不当。
(2) 由于取样系统潜在的粒子损失,粒子在低浓度和粒径大于1μm 时样品收集的局限性,使得该粒径下的洁净度级别不适用。
(3) 为了规定ISO 5 级的粒径,大粒子M 描述符可能适用于至少一种其他粒径联合使用。
通过对分级确认过程中不同标准所参考的粒径以及浓度进行对比分析,其中欧盟GMP 附录1 中的分级确认要求与ISO 14644-1 2015(E)保持一致,与我国确认时的要求存在差距。为了满足不同客户销售市场法规的要求,建议在进行确认时,同时对0.5μm、1.0μm 和5.0μm 进行监测。
3.2 微生物
3.2.1 中国GMP2010 版
中国GMP2010 版附录1 中对于“动态”确认过程中微生物的要求。中国GMP 附录1 中微生物的控制标准见表7。

表7 中国GMP 附录1 中微生物的控制标准Table 7 Chinese GMP Appendix 1 microbiological control standards
由表7 可以看出以下2 点。
(1) 表中各数值均为平均值。
(2) 单个沉降碟的暴露时间可以少于4 h,同一位置可使用多个沉降碟连续进行监测并累计计数。
3.2.2 中国药典2020 版9205
中国药典2020 版9205 对于“动态”确认过程中微生物的要求。中国药典2020 版9205 中微生物的标准见表8。

表8 中国药典2020 版9205 中微生物的标准Table 8 Microbial standards in The Chinese Pharmacopoeia 2020 Edition 9205
由表8 可以看出以下2 点。
(1) 表中各数值均为各取样点的测定值。
(2) 单个沉降碟的暴露时间可以少于4 h,同一位置可使用多个沉降碟连续进行监测并累计计数。如果试验时间少于4 h,则仍应使用表中的限度。
3.2.3 欧盟GMP2020 年附录1 草案
欧盟GMP2020 年附录1 草案无菌药品中则提出了新的标准。洁净室的微生物浓度应为洁净室确认的一部分。采样地点的数量应基于文件化的风险评估,评估依据包括洁净室分级结果,气流流行研究以及对该区域要执行的过程和操作的了解。确认需要在静态和动态分别进行。欧盟GMP 无菌附录1 中微生物的标准见表9。

表9 欧盟GMP 无菌附录1 中微生物的标准Table 9 European Standards for microorganisms in Appendix 1 of GMP sterility
由表9 可以看出以下2 点。
(1) 欧盟附录1 中明确要求A 级区的微生物不得生长,中国GMP2010 版附录1 和中国药典2020 版中均为<1;其中中国GMP 和中国药典2020 版表格中要求的内容描述不同,药典中要求的数值为测定值,GMP 中为平均值,这样理解,药典规定实际上和欧盟GMP 附录一致,不得生长,只是表述方式不同。而GMP 中所要求的可能会存在,当某取样点放置多个取样碟时,在进行均值计算时会掩盖微生物生长的现象。
(2) 各国法规中均明确了动态的测试标准,静态标准。
可参照动态标准升一个级别,如C 级动态沉降菌为50cfu/4 h,静态可设置为B 级的动态5cfu/4 h。
根据确认过程中的历史数据。
和动态标准保持一致。
需进行空调系统确认的条件有以下4 条。
(1) 当洁净厂房和空调系统发生变更或较大的维修后,可能对洁净区的环境产生影响时,需重新进行确认。
(2) 法规中有明确规定的需要定期进行再确认的项目。
(3) 在日常的环境监测过程中,发现连续超出行动限,且经分析是由于系统原因所致,需对系统进行再确认。
(4) 停工时间较长,且期间未对系统进行预防性维护检修,再次使用前,需对系统进行再确认。
4 日常环境监测过程中的取样点和位置
4.1 环境监测静态及动态要求和取样点位置和数量
日常环境是否进行静态和动态监测及环境监测取样点的位置和数量的确定原则如下。
(1) 中国GMP 2010 版无菌附录1:第三章第十条,应当按以下要求对洁净区的悬浮粒子进行动态监测;第十一条,应当对微生物进行动态监测,评估无菌生产的微生物状况。
(2) 欧盟2020 年2 月无菌附录1(草案):9.5 条洁净区、洁净空气设备和人员的日常监测应在动态条件下贯穿所有关键阶段,包括设备装配。
(3) 无菌工艺模拟试验指南(无菌制剂)2018 年10 月,其中6.12.1 环境监控方案设计中给出了如下要求:应通过风险评估确定日常生产环境监控的各要素,如取样点、取样对象、取样频率、警戒和纠偏标准、实施方法等。
(4) USP<1116>中也有相关的描述:监测点应当根据风险评估确定。
(5) 欧盟2020 年2 月无菌附录1(草案):9.4 为了建立一个全面的环境监测方案,应进行风险评估,即采样地点、监测频率、使用的监测方法和培养条件(例如时间、温度、有氧和/或厌氧条件)。
通过以上法规、规范的描述看出,日常环境监测时没有对静态监测进行特殊要求,其中动态监测时取样点和取样位置需根据风险评估的结果确定。
4.2 如何对取样位置和取样点数量进行风险评估
根据产品的生产工艺流程,确定在每个房间内所进行的生产活动,房间的洁净等级,生产操作细节和具体的操作流程,持续的时间,对产品可能造成的污染方面(悬浮粒子和微生物)。
4.2.1 房间风险等级分类
根据房间的洁净等级和生产活动性质确定目标房间的污染等级,一般分为严重、高、中和低。
房间风险等级列表见表10。房间评估示例分析见表11。

表10 房间风险等级列表Table 10 List of room risk levels
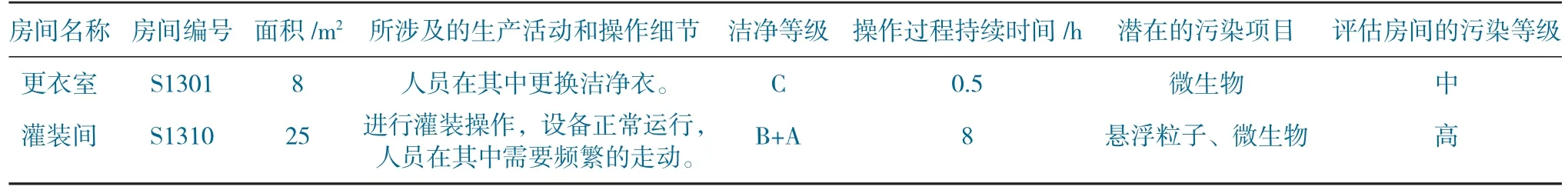
表11 房间评估示例分析Table 11 Room evaluation sample analysis
4.2.2 关键监测点的确定原则
针对风险评估为高、中的房间,需确定房间内的关键监测点,选择实施动态监测或连续监测。监测点的数量需包含所有的风险区域。监测点的评估要求列表见表12。

表12 监测点的评估要求列表Table 12 List of assessment requirements for monitoring sites
监测点评估示例分析见表13。

表13 监测点评估示例分析Table 13 Sample analysis of monitoring point evaluation
4.2.3 监测方式和频次的确定
根据确定的风险分级情况,采取相应的监测方式和频次。定义监测方式和频次见表14。
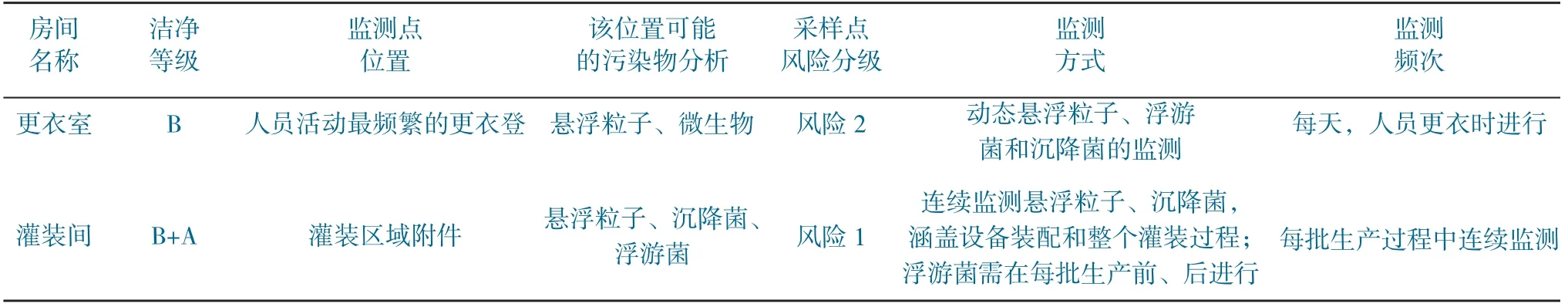
表14 定义监测方式和频次Table 14 Define monitoring mode and frequency
5 结 语
洁净区、空调系统确认过程和日常的环境监测属于2 个不同的阶段。其中确认过程是对系统在静、动态(模拟最差条件) 条件下,是否能够满足生产过程所要求的洁净级别的测试过程,确定系统的能力,并形成文件化证据。而环境监测,是监控生产过程中环境的洁净度是否能够满足产品生产的要求,从而保证所生产的产品安全、有效,满足患者的要求。这是一个最直观的因素,为产品的放行提供了有力的数据支持。本文通过对比这2 个阶段国内、外相关法规中的要求,提出了对于确认需根据市场需求选择合适的采样点数量和位置,而对于日常环境监测采样点的位置和数量需根据风险评估确定,风险评估的方式也在文中进行了相应的描述,给医药行业的实施提供了参考。
