以橘子皮为碳源合成的CeO2@C及其对酸性染料去除性能
2022-03-16肖忠连伍轩逸谭贺云郝仕油
肖忠连 伍轩逸 谭贺云 郝仕油
(浙江师范大学行知学院,生化学院,金华 321004)
众所周知,环境污染是人类面临的棘手问题。有机染料废水是导致环境污染的主要原因之一,因为这些染料含有致癌、致畸、致突变的三致有机污染物。我国是染料生产大国,目前各种染料生产总量已超过90万吨,占全世界的60%,其中2%左右的染料在生产过程中流入水体中,造成严重的环境污染。因此,当务之急是采用合理方法减少有机污染物排放,降低其在环境中的含量。
研究表明,光催化技术是行之有效的方法,因为在光催化过程中,会产生具有氧化性的物质如超氧自由基[1]和羟基自由基[2],对有机染料进行降解,产生无毒、无害的小分子物质如水和二氧化碳。目前研究较多的半导体为TiO2及其改性复合物。TiO2具有无毒、合成工艺相对简单、对紫外光具有较强吸收等优点,因而在光催化方面具有较广泛的应用[3-5]。然而,TiO2对可见光吸收较少,从而限制其在实践中的应用[6]。因此,寻求其它半导体势在必行。由于CeO2对可见光具有潜在吸收,近年来以其为光催化剂对有机污染物进行降解的研究日益增加[7-9]。为了提高对可见光的吸收及对有机污染物的吸附进而提高光催化效率,不同类型的碳物种(活性炭、石墨烯、C3N4)与CeO2的复合物应运而生[10-11]。以上碳材料与CeO2复合后,虽然可提高CeO2的光催化效率,但它们都是工业成品,存在使用成本较高、碳与CeO2间成键率较低等问题[12]。因此,寻找可替代碳源迫在眉睫。最近,Qian等以豆芽为原料,合成了CeO2碳量子点复合物,研究结果表明该物质的光催化产氢效率明显高于相应催化剂[13]。除了以豆芽为碳源,还能利用其它更便宜且易得的生物质为碳源,制备具有较高光催化效率的CeO2@C复合物吗?
研究表明,橘子皮主要由纤维素、果胶、半纤维素、木质素、叶绿素等含羟基、羧基官能团的低分子量碳化物组成,通常是果汁和软饮料工业中的废弃物,过度积累会造成土地占用,产生有污染的酚类化合物。有研究报道,可通过一定的化学方法对橘子皮进行改性,获得多孔结构,提高羟基和羧基相对含量,进而提高其对有机污染物的吸附能力,使其成为吸附载体或光催化辅助剂对污水进行处理[14-15]。橘子皮是易得的生物质,除具有上述官能团及功效之外,还是重要的碳源。基于此,本研究以橘子皮为碳源,合成了CeO2@C复合物,并对其光催化性能进行了研究,结果表明该复合物对废水中的酸性染料具有良好的光催化效率。
1 实验部分
1.1 药品与材料合成
六水合硝酸铈(Ce(NO3)3·6H2O)、氢氧化钠、37%盐酸、28% NH3·H2O、无水乙醇、碘化钾(KI)、对苯二甲酸(PTA)均为分析纯,购自上海国药试剂有限公司。酸性橙7(AO7,AR)购自南京化学试剂股份有限公司。对苯醌(BQ,AR)购自上海谱振生物科技有限公司。橘子皮为从浙江金华市场上买来的橘子去果肉后获得。
取50 g橘子皮,将其用去离子水清洗几次,除去表面灰尘和其他污染物,在60℃恒温鼓风干燥箱内干燥24 h。烘干后取出,放入500 mL烧杯中,向其中加入200 mL无水乙醇,除去橘子皮中的橙皮甙、香精油和色素等小分子化合物)、200 mL 0.1 mol·L-1的NaOH溶液,充分浸泡72 h后水洗至中性,放入60℃恒温鼓风干燥箱。24 h后取出研磨成粉末,制得橘子皮粉末(OPP)。
分别称取3份0.43 g Ce(NO3)3·6H2O放入100 mL烧杯中,向烧杯中加入50 mL去离子水,持续搅拌5 min后向其中分别加入0.1、0.2、0.3 g OPP,继续搅拌30 min使OPP充分分散在Ce(NO3)3·6H2O溶液中。逐滴滴加 4 mL NH3·H2O,继续搅拌 10 min,隔绝空气放置6 h后抽滤,再用去离子水、无水乙醇各洗3遍,放入60℃烘箱干燥12 h,即得CeO2·xH2O@OPP,根据加入的OPP质量依次命名为CeO2·xH2O@OPP-1、CeO2·xH2O@OPP-2、CeO2·xH2O@OPP-3。在相同实验条件下,不添加OPP时制得CeO2·xH2O。
分别称取CeO2·xH2O@OPP-1、CeO2·xH2O@OPP-2、CeO2·xH2O@OPP-3 各0.1 g,放入石英方舟内,再把石英方舟放入管式炉中,通入N2驱除O2,以5℃·min-1的升温速率升温至600℃,煅烧3 h,制得样品,分别命名为CeO2@C1、CeO2@C2、CeO2@C3。在相同实验条件下,煅烧未复合OPP的CeO2·xH2O即获得CeO2。
1.2 样品表征
红外测试在Nexus670型傅里叶变换红外光谱仪上进行,测试条件:光谱范围4 000~650 cm-1,分辨率4 cm-1,扫描累加次数128次。X射线衍射(XRD)图在PW3040/60型X射线衍射仪上获得,工作条件为Cu Kα(λ=0.154 06 nm),40 kV,40 mA,扫描速度6(°)·min-1,扫描区间 20°~80°。Mapping分布图在日立公司S-4800场发射扫描电镜(FE-SEM)上获得,工作电流为10 mA,加速电压为5 kV。Raman光谱在Renishaw公司RM1000型显微共聚焦激光拉曼光谱仪上获得,激光波长(He-Ne,约3 mW)为632.8 nm,扫描范围为200~700 cm-1。UV-Vis光谱在Nicolet Evlution 500双光束型光谱仪上获得,扫描速率为240 nm·min-1,扫描范围200~800 nm。X射线光电子能谱(XPS)实验在Perkin Elmer公司PHI-5000 C ESCA型仪器上进行,工作电压为250 W,以Al Kα(E=1 486.6 eV)为激发源。光电流测试在北京完美光源公司PLS-SXE300/300UV型电化学工作站上进行,以Ag/AgCl为参比电极,Pt电极为对电极。光催化后溶液中总有机碳量(TOC)在XPERT-TOC/TNb总有机碳分析仪上进行,采用TEIS分析控制软件实现仪器全自动化操作。
1.3 吸附实验
分别称取含不同OPP量的CeO2·xH2O@OPP各0.04 g,置于40 mL浓度为500 mg·L-1、初始pH值为5.23的 AO7溶液中,磁力搅拌,分别在1、3、6、10、20、40、60、90、120 min处取样5 mL,离心分离溶液和吸附剂,用UV-2501PC型紫外可见光谱仪测定离心后的上清液(4 mL)在484 nm处的吸光度,根据公式1计算吸附量:

式中Qt(mg·g-1)为t时刻吸附量,若此时达到平衡,则为平衡吸附量Qe(mg·g-1);c0(mg·L-1)和 ct(mg·L-1)分别为吸附前和t时刻染料的浓度;V(mL)为溶液的体积;m(g)为吸附材料的质量。
1.4 光催化实验
取 40 mg光催化剂(CeO2、CeO2@C),放入 40 mL浓度为0.2 mmol·L-1的AO7溶液中,暗反应30 min,以达到吸附-脱附平衡;然后向其中滴加1 mL 30%的 H2O2,用 300 W 卤钨灯(λ=380~820 nm)作为可见光源照射溶液,分别在不同时间下取样,取样间隔时间为 5、15、15、15、15 min。取上层清液(5 mL)离心,在AO7最大吸收波长484 nm处检测吸光度,根据式2计算AO7降解率:

式中c0′、c分别为光催化前和t时刻AO7溶液的浓度,可根据朗伯-比尔定律A=εbc(ε:吸光系数,b:比色皿宽度,c:AO7溶液浓度)计算。
为了进一步研究CeO2@C催化剂的循环利用价值,我们对催化剂进行5次重复实验,实验过程中会因操作问题导致部分催化剂损失,可通过添加相应量的CeO2@C以保证质量相同。重复实验过程除了水洗、烘干之外,其他实验操作与上述光催化过程相同。
2 结果与讨论
2.1 样品结构和性能表征
为了探究 CeO2·xH2O 及 CeO2·xH2O@OPP 表面官能团情况,我们对材料进行了FT-IR分析,结果如图1所示。由图可知,所有材料在1 037、1 380、1 624、2 925、3 423 cm-1处均有明显的红外峰。这些峰的归属如下:479 cm-1处为Ce—O拉伸振动峰[16];1 624和1 037 cm-1附近的峰分别归属于离子化羧基(—COO-)中C=O键的对称伸缩振动和多糖的—C—O—C—振动[17];1 380 cm-1附近的峰归因为制备过程中残存的NO3-[18];2 925 cm-1处观察到的峰可认为是CH、CH2和CH3中的C—H伸缩振动所致[19];3 423 cm-1处宽而强的吸收峰对应于OPP中纤维素、果胶、木质素以及材料表面吸收水的O—H拉伸振动[17]。所有含 OPP 的材料,在 1 037、1 624、2 925、3 423 cm-1处的峰有所加强,且随OPP含量增加峰强度逐渐增加,而在1 380 cm-1处的峰逐渐降低,表明OPP与CeO2成功复合。

图1 CeO2·xH2O、CeO2·xH2O@OPP-1、CeO2·xH2O@OPP-2、CeO2·xH2O@OPP-3的FT-IR谱图Fig.1 FT-IR spectra of CeO2·xH2O,CeO2·xH2O@OPP-1,CeO2·xH2O@OPP-2,CeO2·xH2O@OPP-3
图2为CeO2、CeO2@C1、CeO2@C2和CeO2@C3的XRD图。可以看出,上述4种材料的XRD图在2θ为28.5°、33.0°、47.4°、56.3°、59.1°、69.4°、76.7°、79.1°处均出现了明显的衍射峰,分别对应(111)、(200)、(220)、(311)、(222)、(400)、(331)、(420)晶面,与 CeO2标准卡片(PDF No.34-0394)基本一致,表明它们都具有典型立方萤石相结构。图中没有出现其他杂峰,表明OPP在陈化结晶过程中并未影响CeO2的晶形;此外,在图中并未发现碳的衍射峰,证明碳以非晶态形 式 存 在 。 对 比 CeO2、CeO2@C1、CeO2@C2 和CeO2@C3的衍射峰可知,随着OPP负载量增大,样品衍射峰强度逐渐减弱,可能原因是CeO2·xH2O@OPP煅烧后,表面包覆的非晶态炭层厚度逐渐增大,使XRD峰强降低。

图2 CeO2、CeO2@C1、CeO2@C2、CeO2@C3的XRD图Fig.2 XRD patterns of CeO2,CeO2@C1,CeO2@C2,CeO2@C3
图3是CeO2·xH2O@OPP-1和CeO2@C1的FT-IR谱图。据图可知,CeO2·xH2O@OPP-1经高温煅烧后,在1 054、1 612及2 922 cm-1附近的峰强度基本保持不变,证明在CeO2@C1中,原有样品中有机官能团基本未损失。然而,CeO2@C1中位于1 368 cm-1附近的峰强度明显减弱,证明样品中NO3-含量降低,可能原因是高温煅烧使硝酸根分解。CeO2@C2和CeO2@C3的红外峰与CeO2@C1的类似。

图3 CeO2·xH2O@OPP-1和CeO2@C1的FT-IR谱图Fig.3 FT-IR spectra of CeO2·xH2O@OPP-1 and CeO2@C1
图4为CeO2与CeO2@C1在1 000~2 000 cm-1间的Raman谱图。从图中可以看出,CeO2@C1在1 200~1 800 cm-1区间有2个峰,其中位于1 371 cm-1附近的D带,是由无序石墨结构或缺陷干涉带边(k点附近)声子所致;而位于1 618 cm-1处的G带与碳材料中sp2原子的面内振动有关[20]。D带和G带也同样存在于CeO2@C2和CeO2@C3的Raman谱图中,证明合成样品中有碳存在,且碳与CeO2基体形成较强的结合力。为了对比,对纯CeO2进行Raman测试,证明无碳的相关峰出现(图4)。为了进一步证明碳的存在及合成样品中各元素的分布情况,本研究利用能量色散X射线光谱(EDS)技术对CeO2@C1中Ce、O、C进行Mapping测试,结果见图5。可以看出,C与Ce、O相间分散于合成样品中,且各元素均匀分布。其它样品中Ce、O、C分布情形与CeO2@C1中的类似。

图4 CeO2@C1和CeO2的Raman谱图Fig.4 Raman spectra of CeO2@C1 and CeO2

图5 CeO2@C1中各元素Mapping图Fig.5 Elemental mappings of CeO2@C1
不同的晶粒尺寸和结晶度会导致CeO2光学性能发生变化。为了探究碳负载后CeO2的光学性能,我们对煅烧后的样品进行了UV-Vis分析,结果见图6。由图 6 可知,在紫外区(<400 nm)CeO2及所有CeO2@C的漫反射光谱中有一个强的吸收带,可能是O2p和Ce4f轨道之间的电荷转移跃迁所致[21]。对比CeO2@C1与CeO2可以发现,CeO2@C1在紫外区和可见光区的吸收均有所增强;随着前驱体中OPP含量增加,相应煅烧样品在可见光区的吸收提高,可能原因是煅烧后形成的碳量逐渐增大,增强了对可见光的吸收。然而,随着碳量增加,包覆在CeO2表面的碳量厚度逐渐增大,导致样品粒径增大、比表面积减小,因而CeO2@C2和CeO2@C3在紫外区的吸收低于 CeO2和 CeO2@C1的[22]。
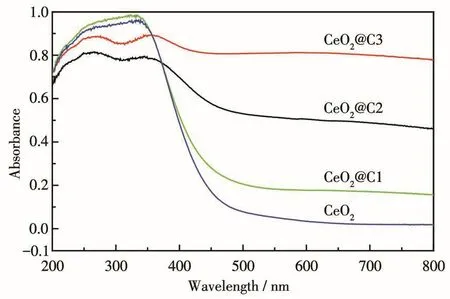
图6 CeO2和不同含碳量CeO2@C的紫外可见漫反射谱图Fig.6 UV-Vis diffuse reflectance spectra of CeO2and CeO2@C with different amounts of carbon
2.2 样品光催化性能测试
为了考察合成样品的光催化性能,本研究以AO7 为 探 针 染 料 ,比 较 CeO2@C1、CeO2@C2、CeO2@C3 和 CeO2对 0.2 mmol·L-1的 AO7 降解效果,结果见图7。据图可知,光催化反应50 min后,CeO2、CeO2@C1、CeO2@C2和 CeO2@C3对 AO7的降解率分别为24%、97%、86%、45%,可见CeO2@C1对AO7的光催化降解效果最好,可能原因是CeO2@C2和CeO2@C3中碳层厚度过大,影响染料吸附效率,导致光催化效率降低。从暗反应30 min结果可知,CeO2、CeO2@C1、CeO2@C2和 CeO2@C3对 AO7的吸附效率分别为39.6%、57.6%、20.7%、10.9%,其中CeO2@C1吸附效果最好。为了进一步证明产物外层包裹物厚度对染料吸附效率的影响,本研究测试了 CeO2·xH2O@OPP-1、CeO2·xH2O@OPP-2 和 CeO2·xH2O@OPP-3对AO7的吸附效率,结果见图8。可见,随着CeO2·xH2O外层OPP含量增加,合成产物对AO7的吸附效率逐渐降低,证明适量包裹外层物有利于合成产物吸附效率的提高。虽然CeO2@C2和CeO2@C3对可见光的吸收强度大于CeO2@C1的(图6),但他们对染料的吸附效率均低于CeO2@C1的,而染料在催化剂表面吸附效率是影响光催化降解效率的重要因素,因此CeO2@C1光催化效率最好。此外,本研究还利用0.05 g OPP合成了CeO2@C0,并对其进行了光催化实验,结果表明,暗反应30 min后,产物对AO7的去除率为46%;光催化50 min后,AO7的降解率为92%;均低于使用0.1 g OPP为碳源所合成产物的相关数据(图7),进一步证明上述结论。对比CeO2和CeO2@C的光催化降解率可知,引入碳后,CeO2对AO7的吸附及光催化效率均有所提升,较好的光催化效率是因为提高了产物对可见光及染料吸附效率,还可归因于碳对光生电子的高效捕获作用[23]从而使光生电子与空穴高效分离。为了验证橘子皮来源对产物光催化效率的影响,本研究还采用产自江西九江的橘子皮进行实验,结果表明橘子皮产地不同对产物光催化效率基本无影响,详见Supporting information中的结果与讨论。

图7 CeO2及含不同碳含量CeO2@C对0.2 mmol·L-1 AO7的光催化活性Fig.7 Photocatalytic activity of CeO2and CeO2@C with different amounts of carbon for 0.2 mmol·L-1of AO7

图8 CeO2·xH2O@OPP-1、CeO2·xH2O@OPP-2和CeO2·xH2O@OPP-3对AO7的吸附效率Fig.8 Adsorption efficiency of AO7 on CeO2·xH2O@OPP-1,CeO2·xH2O@OPP-2,and CeO2·xH2O@OPP-3
为了探究光催化过程中AO7褪色是由于光催化还是吸附所致,本研究对光催化后的材料进行回收(回收之后的材料用去离子水彻底清洗,在60℃下烘干),并进行FT-IR测试。图9是CeO2@C1、回收之后的CeO2@C1及AO7的FT-IR谱图。可以看出,光照之后的催化剂与原催化剂的红外峰除峰强度有所降低外,其他均无变化;回收之后的CeO2@C1的FT-IR谱图中未出现AO7的特征峰。因此,可推断AO7褪色是由于CeO2@C1对其进行光降解所致。为了验证此推论,本研究对不同光催化时间下溶液中TOC去除率进行计算,结果如下:70%(5 min)、88%(20 min)、92%(35 min)、93%(50 min)、94%(65 min),与图7中CeO2@C1光催化降解结果基本相同,证明AO7褪色确实因降解所致。

图9 CeO2@C1、回收的CeO2@C1和AO7的FT-IR谱图Fig.9 FT-IR spectra of CeO2@C1,the regenerated CeO2@C1,and AO7
为了深入理解CeO2@C1对AO7的光催降解机制,本研究采用XPS对合成样品的表面组成和化学状态进行分析,结果详见图10。据图10a可知,样品中存在 Ce3+和 Ce4+,因为 903.0 eV(U′)和 884.9 eV(V′)处的分峰可归属于 Ce3+[24],而在 916 eV(U‴)、907.1 eV(U″)、900.4 eV(U)、897.8 eV(V ‴)、888.4 eV(V″)和881.8 eV(V)处的分峰与Ce4+有关[25]。据以往研究可知,当Ce3+存在时,它可取代Ce4+,导致氧空位产生[12]。据图10b可知,氧空位确实存在于CeO2@C1中,因为O1s中530.8 eV处分峰是缺陷氧所致,而528.8 eV处分峰为晶格氧所致[25]。据图10c可知,在CeO2@C1中存在较多的碳键(C=C、C—C、C—OH、C=O、O—C=O键),类型多于以活性炭为碳源合成的CeO2@C[12]。较多的碳键有利于光生电子传导,利于其与空穴分离,因而光催化效率提高。由于形成较多的碳键,所以CeO2@C1的光生电流强度大于CeO2(图11),因而其光催化效率高于CeO2;此外,CeO2@C1中形成的氧空穴易捕获光生电子[26],提高光生电子与空穴分离效率,且可通过静电引力(氧空位带正电,AO7中磺酸基带负电)作用提高AO7的吸附量,导致CeO2@C1光催化效率提高。

图10 CeO2@C1中Ce3d(a)、O1s(b)和C1s(c)XPS谱图Fig.10 XPS spectra of Ce3d(a),O1s(b)and C1s(c)for CeO2@C1

图11 CeO2@C1和CeO2的光电流-时间曲线Fig.11 Photocurrent-time profiles of CeO2@C1 and CeO2
为了进一步理解光催化机理,本研究采用KI(用于检测h+)、BQ(用于检测·O2-)、PTA(用于检测·OH)检测催化过程中的活性物种,结果详见图12。据图可知,添加PTA对光催化效率基本不影响,证明·OH不是活性物种;而加入KI后,开始时对降解效率有一定影响,但随着时间增长,光催化效率受影响程度不大;但是加入BQ后,极大地降低了AO7的降解率,证明·O2-是主要的活性物种。此外,我们还研究了H2O2对CeO2@C1光催化效率的影响,结果详见图13。可以看出,H2O2可提高CeO2@C1的光催化效率,可能原因是氧空位可提高H2O2的吸附,而吸附H2O2可促进 Ce3+和Ce4+的循环,伴随 O2产生(式 3),当O2结合光生电子后就生成·O2-,从而降解 AO7[27]。另外,从循环实验结果可知,所合成的CeO2@C1稳定性较高,因为循环使用5次后,其光催化效率仅降低9%左右。

图12 CeO2@C1光催化活性物种捕获实验Fig.12 Trapping experiment of photogenerated reactive species for CeO2@C1
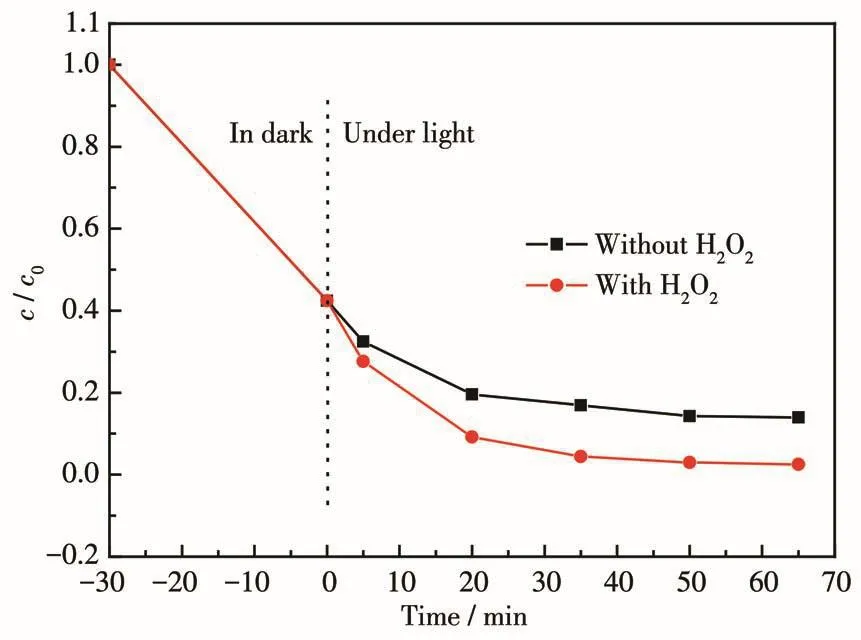
图13 H2O2对CeO2@C1光催化降解0.2 mmol·L-1AO7活性的影响Fig.13 Effect of H2O2on photocatalytic activity of 0.2 mmol·L-1AO7 over CeO2@C1
3 结 论
以处理过的橘子皮为碳源合成了CeO2@C复合物。光催化结果表明,适量引入碳源可较大幅度提高产物光催化效率。由于碳均匀分布于合成样品表面,导致产物对可见光吸收效率提高,同时由于在产物内形成了较多氧空穴及碳键,利于光生电子与空穴分离,因而光催化效率大幅度提升。由于合成样品光催化效率及稳定性较高,CeO2@C1有望在实践中成为处理其它有机污染物的光催化剂。
Supporting information is available at http://www.wjhxxb.cn
