唾液菌群的直接PCR 扩增-高分辨率熔解曲线分析及其法医学应用
2022-03-15杨瑞陈炯赵贵森
杨瑞,陈炯,赵贵森
河南科技大学法医学院,河南 洛阳 471023
细菌是人体的重要组成部分,在人体各部位形成复杂多样的微生态系统[1-2]。由于性别、年龄、生活环境、饮食习惯、健康状况等的不同,人体菌群具有个体特异性,同一个体不同部位的菌群也不相同[3]。人体菌群很容易转移到现场物品上,或随分泌物、排泄物遗留在现场,甚至比人体细胞的数量还多,且能在一定时间内保持菌群原有的特性[4]。人死后,尸体各处的菌群随腐败进展而发生有规律的变化[5-6]。现场检材或尸体的菌群特征可提供斑痕类型、来源个体及死亡时间等信息[7-8],菌群分析作为解决法医学问题的新途径日益受到关注。
菌群可用形态、生物化学和遗传学等方法进行分析,但受检材条件等限制,目前法医学菌群分析主要指菌群的DNA 分析,常用的标记基因是以序列变异为主的16S rDNA,分析过程一般包括检材处理、DNA提取、扩增、PCR 产物检测和数据分析等多个环节[4-10]。在DNA 提取环节,又有裂解、纯化等步骤,耗时长,检材用量大。PCR 产物则须用杂交、测序或梯度凝胶电泳等特殊的技术进行分离、检测,操作繁琐,成本高,污染风险大。此外,菌群PCR 是一种以群体特征为研究对象的多模板PCR[11],不同于法医学常用的多重PCR,不能按照传统的PCR 来优化和评价。现有的菌群分析技术均来自其他领域,不符合法医学工作特点,也未经改造和验证,难以满足法医学检验的要求。
熔解曲线分析法利用解链温度差异来鉴别DNA,在荧光定量PCR 仪上扩增的DNA 可直接进行熔解曲线分析[12]。使用EvaGreen 等饱和荧光染料和高精度的PCR 仪,可以提高熔解曲线的分辨率,即高分辨率熔解(high resolution melting,HRM)。HRM 过程简单、快速,灵敏度和信息含量高,已应用于变异或突变筛查、病原体快速检测、法医DNA 分型等[13-16]。关于HRM 在菌群分析中的应用,EVERMAN 等[17]已有初步探索,WANG 等[18]也已将其用于人类唾液菌群的比较,但都要先提取菌群的宏基因组DNA。直接PCR(direct PCR,dPCR)扩增已是成熟的法医DNA 分型技术,在菌落筛选、病原体诊断中也有应用[19],可以简化操作、缩短时间,是菌群DNA 分析快速化的另一潜在途径。16S rDNA 是菌群分析中最常用的标记基因,其V4 高变区在进化关系上更接近16S rDNA 全长,通用性和分辨率均较高,且在法医学领域已有报道[18,20]。
本研究拟结合dPCR 和HRM 技术,以16S rDNA V4 区为例,建立一种免提取、免电泳的唾液菌群快速检测技术,即dPCR-HRM 技术,并评估其法医学应用价值。
1 材料与方法
1.1 主要试剂和仪器
热启动Taq酶、dNTPs、Ex TaqBuffer(Mg2+free)和MgCl2均购自日本TaKaRa 公司,20×EvaGreen 荧光染料购自美国Biotium 公司,多杀性巴氏杆菌(Pasteurella multocida,Pm)A01 菌株的基因组DNA 由河南科技大学基因工程疫苗课题组赠送,细菌基因组DNA提取试剂盒购自天根生化科技(北京)有限公司,Tris-EDTA(TE)缓冲液(pH=8.0)购自北京索莱宝科技有限公司。细菌16S rDNA V4 区的通用引物[18]由北京博迈德基因技术有限公司合成,序列为:518F(5′-CC AGCAGCCGCGGTAAT-3′)和806R(5′-GGACTACCA GGGTATCTAATCCTGTT-3′)。
Rotor-Gene Q 实时荧光定量PCR 仪(德国Qiagen公司),FLA6000 微量紫外可见分光光度计(杭州晶飞科技有限公司),TGL-16B 高速台式离心机(上海安亭科学仪器厂)。
1.2 样本采集
按照知情同意原则,采集本校79 名志愿者的非刺激性唾液[21]共84 份。采集前至少禁食禁水1 h,采集时志愿者身体前倾,收集自然流出的唾液约1 mL于1.5 mL 微量离心管中并编号,于采样后30 min 内进行后续处理或置于-80 ℃保存备用。其中,1~5 号志愿者为本课题组成员(年龄20~60 岁;男性3 名,女性2 名;吸烟者2 名,不吸烟者3 名),在初次采样3 个月后重复采样1 次,初次采样样本编号为1~5 号,第二次采样样本编号为T1~T5。1 号、2 号志愿者在第二次采集唾液的同时无菌采集外周静脉血,常规Chelex 法提取基因组DNA。其他样本来自本校学生,编号6~79号。
本研究已获得河南科技大学第一附属医院医学伦理委员会批准(审批文号:20180306)。
1.3 实验方法
1.3.1 样本预处理
唾液:新鲜或快速解冻平衡至室温的唾液,涡旋振荡30 s,常温下以离心半径4 cm,3 000 r/min,离心30 s,将上清液移至另一1.5 mL 微量离心管中,以离心半径4 cm,12 000 r/min,离心10 min,弃去上清液,加入等体积TE 缓冲液重悬。
唾液斑:10 号、11 号唾液样本在采集后立即制成两组唾液斑,每组20 份,每份均取10 μL 滴加到载玻片上,室温放置形成唾液斑,分别沉积0、0.5、2、4、8 h后用TE 缓冲液润湿的纱线擦拭转移至加有50 μL TE 缓冲液的微量离心管中,室温浸泡30 min,涡旋振荡30 s,以离心半径4 cm,3 000 r/min,离心30 s,取上清液作为PCR 模板。
混检样本:分别取编号为T1~T5 的唾液样本各50 μL 等体积混合,制成可以代表一般个体唾液细菌含量和菌群结构的混检样本[22],采取与唾液样本相同方法处理后,分为2 份:1 份从3.5 倍至3.5×105倍行10 倍梯度稀释;另1 份用细菌基因组DNA 提取试剂盒提取,紫外分光光度法定量后,从250 ng/μL 至2.5×10-3ng/μL 行10 倍梯度稀释。
1.3.2 菌群宏基因组DNA 提取
取T1~T5 号唾液样本按1.3.1 节方法制成TE 悬液各50 μL,分别用细菌基因组DNA 提取试剂盒提取DNA,具体操作参照试剂盒说明书。用微量紫外可见分光光度计测定DNA 浓度,4 ℃保存备用。
1.3.3 PCR-HRM 检测
PCR 体系为20 μL,包括:10 μmol/L 的正、反向引物各0.4 μL,2.5 mmol/L dNTPs 1.6 μL,10×PCR 缓冲液2 μL,25 mmol/L MgCl22.4 μL,20×EvaGreen 1 μL,5 U/μL 热启动Taq酶0.2 μL,以1.3.1 节制备的细菌悬液或1.3.2节提取的菌群DNA为模板,补纯水至20 μL。循环参数:95 ℃ 10 min 预变性;94 ℃ 10 s,65 ℃ 10 s,72 ℃ 10 s,40 个循环,采用降落PCR,前25 个循环每次退火温度降低0.5 ℃。HRM 条件:95 ℃变性1 min,降温至40 ℃保温1 min,再以最高速度升温至75 ℃后开始以每步0.2 ℃逐步升温、采集荧光信号,直至95 ℃。上述过程即为PCR-HRM。其中,直接以1.3.1 节制备的细菌悬液为模板时,称为dPCR-HRM;以1.3.2 节试剂盒(kit)提取的细菌基因组DNA 为模板时,称为kPCR-HRM。
每批次检测均以多杀性巴氏杆菌的基因组DNA和纯水分别作为阳性和阴性对照模板,模拟斑痕实验时以擦拭载玻片空白区的纱线同步制作空白基底对照。以Chelex 法提取的人外周静脉血基因组DNA 为无菌模板,验证引物和PCR 扩增的特异性。每个反应至少设置两个复管。
1.4 方法评估
以已知序列的阳性对照模板(多杀性巴氏杆菌基因组DNA)优化反应体系、循环参数,建立细菌16S rDNA V4 区的PCR-HRM 法,所得图谱与uMelt[23](https://dna-utah.org/index.html)预测图谱比较,并用10 倍梯度稀释(250 ng/μL 至2.5×10-3ng/μL)的多杀性巴氏杆菌DNA 测试灵敏度。
分别以混检样本的菌群DNA、菌群悬液及其梯度稀释液为模板,优化建立的kPCR-HRM、dPCR-HRM技术,并测试灵敏度。以建立的kPCR-HRM、dPCRHRM 法分别检测T1~T5 号样本及混检样本,比较两种方法所得图谱并验证其可行性和鉴别能力。
使用dPCR-HRM 法,间隔1 d、1 周重复检测T1~T5 号唾液样本,评价方法的稳定性;调查两组模拟唾液斑样本,分析dPCR-HRM 法用于斑痕检材的效果,初步评价其法医学实用性;调查混检样本菌群悬液的梯度稀释液,分析样本稀释对dPCR-HRM 图谱的影响,探索一般个体获得菌群代表性HRM 图谱的稀释度范围;调查61 份唾液样本(编号19~79),根据相似度对样本图谱进行聚类,计数各类样本的例数,评价唾液菌群dPCR-HRM 图谱的群体多样性,再随机从上述61 份样本中抽取6 份进行盲测,与原聚类结果进行比较,验证dPCR-HRM 用于未知样本分型的可行性。
1.5 数据分析
参考NUNZIATA 等[24-25]的方法,以判型置信度(genotype confidence percentage,GCP)为衡量HRM 图谱之间相似性的客观指标。待测样本相对于参考样本 的,其中drt为待测曲线与参考曲线之间的欧氏距离。应用Rotor-Gene Q 2.3.1 软件(德国Qiagen 公司),结合人工核查,进行数据分析和判型。
Rotor-Gene Q 2.3.1 软件采集扩增和熔解过程中的荧光强度数据,自动生成扩增曲线和HRM 曲线。根据扩增曲线的循环阈值(cycle threshold,Ct)排除扩增失败的反应管;然后用“Melt”模式分析,核查熔解曲线的峰型,确定熔解的温度范围(82.5~91 ℃);再用“HRM”模式分析,以熔解前1 ℃(81.5~82.5 ℃)和后1 ℃(91~92 ℃)两个区间的荧光值为准,对原始HRM曲线进行归一化,生成归一化的HRM曲线。在“HRM”模式下,定义“菌群型”及其参考样本,调整GCP 界值,查看待测样本的判型与GCP 值。以GCP=80%为界值[26],判断两个HRM 图谱是否能归为同一种菌群型。复管取均值后,导出GCP 等数据及熔解曲线和HRM图谱。涉及不同批次间的比较,如群体样本调查时,先把上述分批检测、导出的数据用Origin 8 软件(美国OriginLab 公司)进行整合,各批次均包含22 号样本,以控制和修正批次间差异[25],然后导入LabSpec 5软件(日本HORIBA Scientific 公司)统一进行平滑、扣基线和校准,生成与系列图谱相应的数据矩阵,再用LabSpec 5 软件的“Model”模式进行建模分析,结合数据和人工读图判定新检出的“菌群型”,统计各种型的数目。
2 结果
2.1 细菌16S rDNA V4 区的PCR-HRM
经优化反应体系、循环参数,以多杀性巴氏杆菌基因组DNA 为模板,可以得到典型的熔解曲线(图1A)、HRM 图谱(图1B),与根据多杀性巴氏杆菌16S rDNA V4 区序列预测的图谱(图1C~D)基本一致。阴性对照、无菌的人DNA 对照,在超净台内操作时无扩增和熔解信号,但常规条件下循环次数大于35 次时会有非特异性信号。
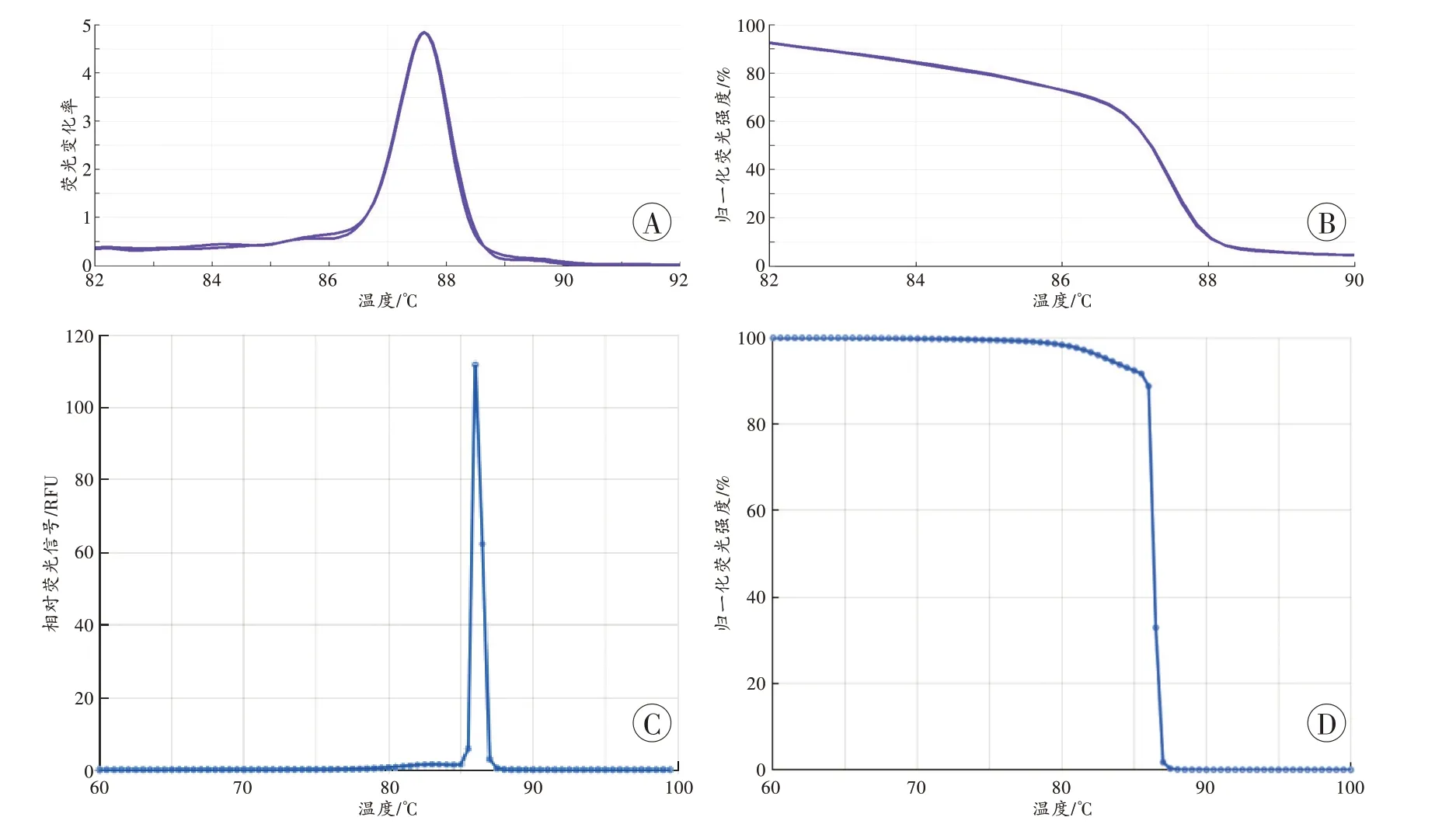
图1 多杀性巴氏杆菌16S rDNA V4 区的PCR-HRM 图谱Fig.1 PCR-HRM profiles of 16S rDNA V4 region of Pasteurella multocida
2.2 菌群的kPCR-HRM
与单一菌株相比,菌群的熔解曲线(图2A)主峰常较宽,有肩峰或多个峰,相应的HRM 曲线(图2B)也可分为多段,各段斜率(即荧光下降速率)不同。
以2.5×10-3ng/μL的混检样本DNA为模板也可得到HRM 图谱,但与该样本模板DNA 量在0.25~25 ng/μL的图谱(图2C)明显不同。以2.5 ng/μL菌群DNA的HRM图谱为参考标准,模板DNA量在0.25~25 ng/μL所得图谱的GCP>96.2%(98.19%±1.75%),而<0.25 ng/μL 或>25 ng/μL所得图谱的GCP<41.98%(32.14%±11.65%)。
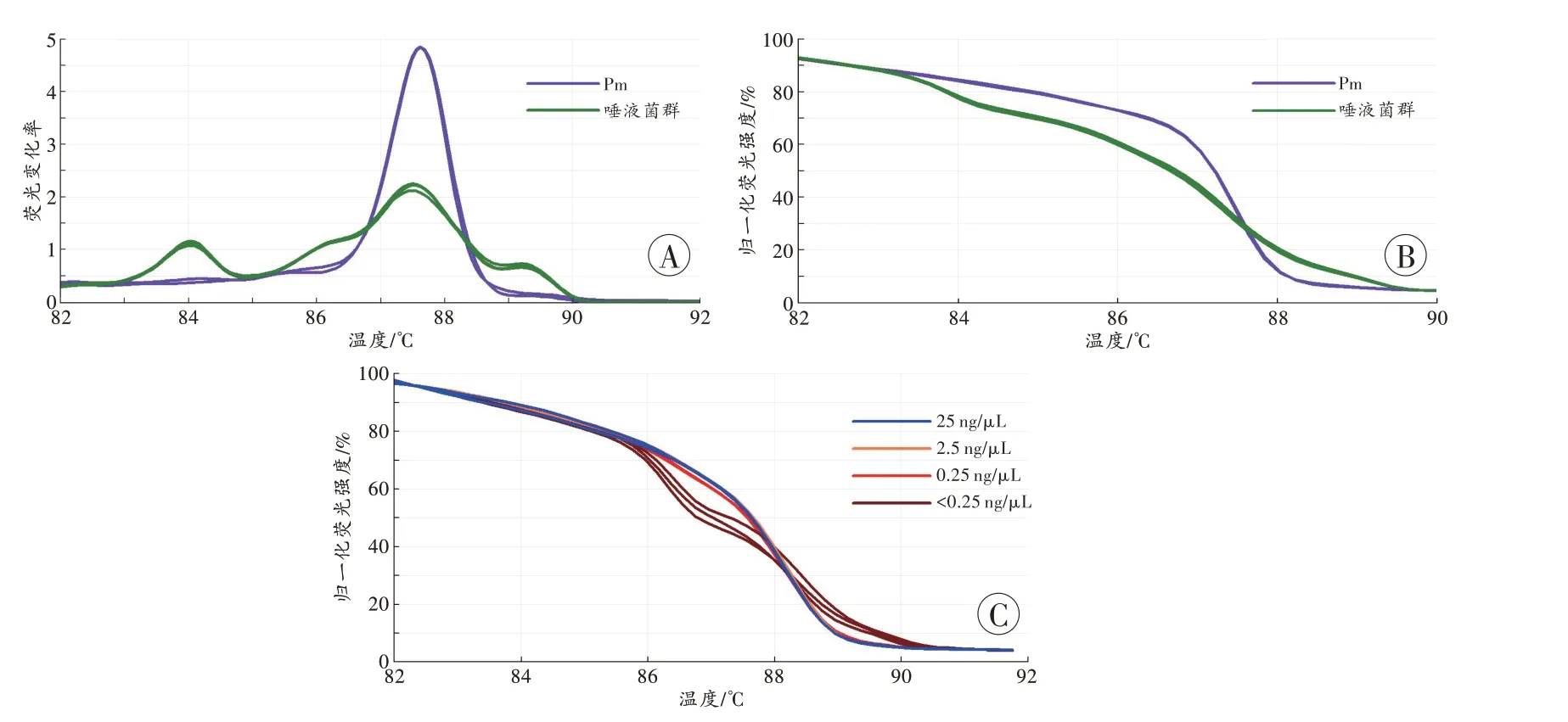
图2 唾液菌群的kPCR-HRM 图谱Fig.2 kPCR-HRM profiles of salivary bacterial community
2.3 菌群的dPCR-HRM
直接以1.3.1节制备的细菌悬液为模板,用dPCRHRM 法进行分析,可以在90 min 内得到样本菌群的HRM 图谱。同一唾液样本,其dPCR-HRM 图谱与kPCR-HRM 图谱相似(图3),两种方法所得HRM 图谱之间的GCP>95.85%(96.95%±1.56%)。
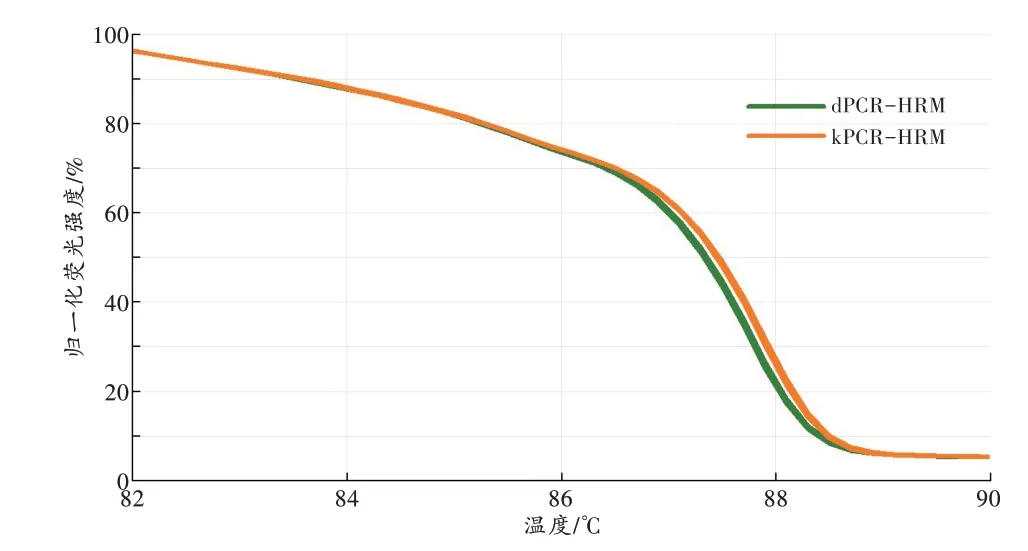
图3 同一样本的dPCR-HRM 和kPCR-HRM 图谱Fig.3 dPCR-HRM and kPCR-HRM profiles of the same sample
稀释35~3.5×103倍的混检样本,模板的唾液量为0.29~29 nL,Ct 值在15~30,dPCR-HRM 图谱基本相似(图4),以350 倍稀释时的图谱为参考标准,GCP>92.11%(96.97%±2.79%)。稀释3.5×105倍,相当于约2.9×10-3nL 的唾液仍可检测,但模板的唾液量大于29 nL 或小于0.29 nL、Ct 值不在15~30 范围内时,HRM图谱可见明显变异(图4),如仍以350 倍稀释时的图谱为参考标准,GCP<81.96%(66.43%±17.23%)。

图4 梯度稀释混检样本的dPCR-HRM 图谱Fig.4 dPCR-HRM profiles of the mixed sample with gradient dilution
2.4 dPCR-HRM 的菌群鉴别能力、稳定性和适用性
用dPCR-HRM 技术检测T1~T5 号唾液样本,不同个体有不同的dPCR-HRM 图谱[GCP<87.07%(52.11%±22.79%),图5]。同一唾液样本,间隔1 d、1周重复检测,所得图谱基本相同[GCP>92.39%(97.78%±2.30%)]。模拟唾液斑样本在纱线转移后使用dPCRHRM 法也可成功检测,且至少在8 h 内图谱相对稳定[GCP>90.83%(96.67%±2.98%),图6]。
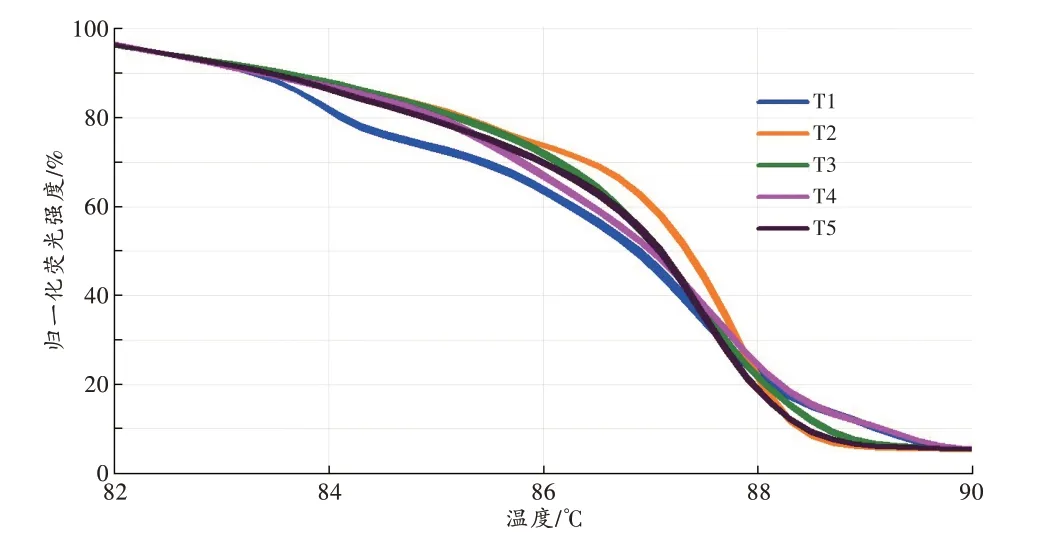
图5 不同个体唾液菌群的dPCR-HRM 图谱Fig.5 dPCR-HRM profiles of salivary bacterial community of different individuals
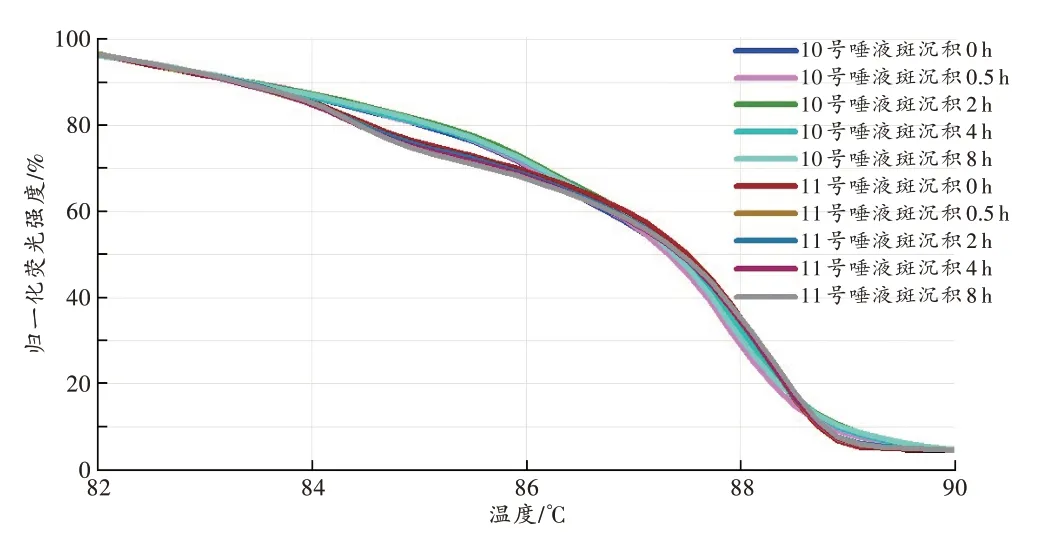
图6 不同沉积时间唾液斑的dPCR-HRM 图谱Fig.6 dPCR-HRM profiles of salivary stains with different deposition times
2.5 群体唾液菌群的dPCR-HRM 型分析
用建立的dPCR-HRM 法集中分析61 名志愿者的唾液,以GCP=80%为界值,可分为10 种基本类型,以①~⑩表示(图7~8)。由①到⑩,各型检出的频数分别为6、3、9、13、2、2、4、9、12、1。再随机抽取6 份进行盲测,除1 个需要修正温度偏离外,对照基本型均可以作出正确判断。
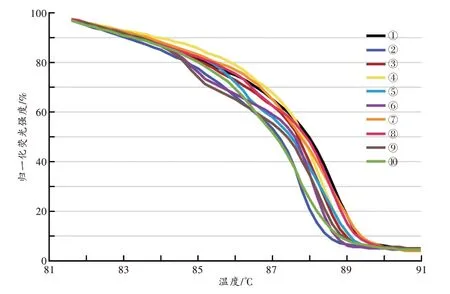
图7 10 种唾液菌群型的归一化HRM 图谱Fig.7 Normalized HRM profiles of 10 bacterial community types in saliva
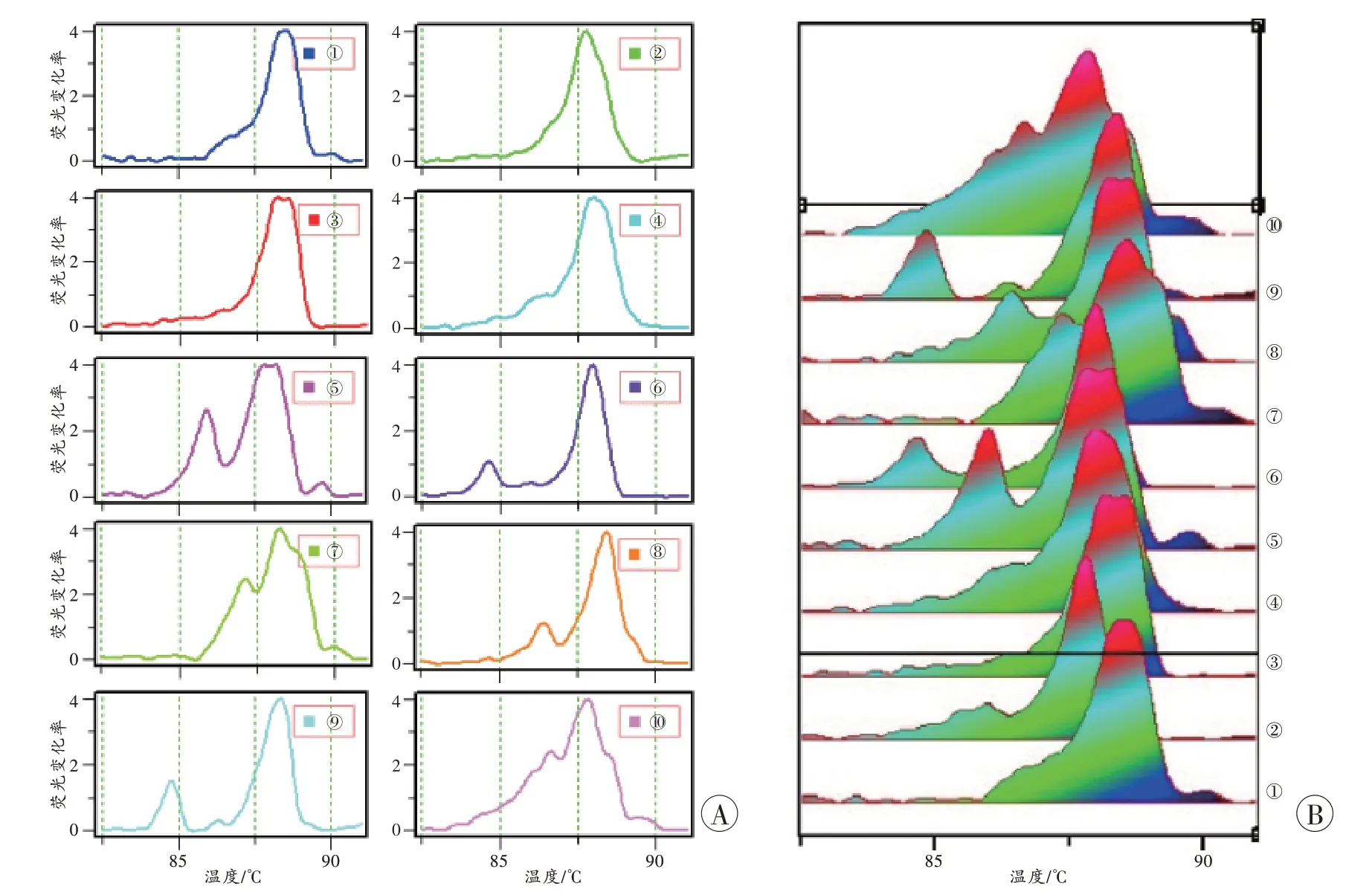
图8 10 种唾液菌群型的熔解曲线Fig.8 Melting curves of 10 bacterial community types in saliva
3 讨论
法医学现场检材中细菌的种类和数量包含着其来源个体、遗留时间等信息,为法医学问题的解决提供了新的思路。但目前的菌群分析技术[27]尚难以满足法医学应用要求,需要结合法医学检验目的、技术要求等进行改进和评价。本研究结合dPCR和HRM 技术建立了一种可用于人体菌群快速分析的dPCR-HRM技术,并初步评价了其应用于法医学领域的可行性。
3.1 dPCR-HRM 菌群快速检测的原理
常规的菌群DNA 分析包括检材预处理、DNA 提取、扩增和PCR 产物分析等,步骤多,耗时长,难以自动化、标准化。其中以DNA 提取和产物分析最为繁琐,耗费检材,且可能改变样本菌群结构,增加污染风险。细菌DNA 的提取有释放和纯化两个环节,即先用机械、理化或酶解等手段破坏细胞,释放DNA,然后通过抽提或吸附等去除其中杂质。热解法不需要特别的设备、试剂,在各种破坏细胞壁的手段中最为简单。唾液等样本经简单处理后可直接加入PCR 体系,利用扩增前预变性时的高温裂解细菌,即可释放出足够的模板[19,22]。研究[28]表明,在基于16S rDNA 的PCR 分析中,各种模板提取方法对结果影响不大,与采样等其他因素相比较几乎可以忽略不计。dPCR 技术本身在法医学领域就应用广泛[29-30],如能实现基于dPCR 的菌群DNA 分析,不仅符合法医学检材特点和检验要求,也更易于普及推广。
菌群检测在PCR 后须用变性梯度凝胶电泳(denaturing gradient gel electrophoresis,DGGE)等较为复杂的方法进行分析,耗时费力[9-11]。双链DNA 的熔解曲线因长度、序列和GC 含量而异,HRM 利用解链温度差异鉴别DNA,为菌群PCR 产物的分析提供了一种新途径。可在扩增前将EvaGreen 荧光染料加入PCR 体系,连续进行扩增和PCR 产物分析。菌群的16S rDNA 片段扩增后,体系内既有来自各种细菌同一基因但序列各异的同源双链DNA,也有这些序列相互杂交形成的、不同程度错配的异源双链DNA。这些同源、异源双链DNA 的解链温度各不相同,拷贝数也因样本的菌群结构而异,整个体系在热变性时会生成复杂的熔解曲线,具体形态取决于样本的菌群结构,即细菌的种类和各种细菌的丰度。HRM 软件把每个样本的曲线作为一个整体进行分析、比较,计算样本曲线之间的欧氏距离,转换成GCP,以客观、量化的形式反映两个样本HRM 曲线的相似度。
联合应用dPCR 和HRM 技术,使用荧光定量PCR仪对样本的菌群进行PCR-HRM 分析,可集传统的提取、扩增和PCR 分析于一体,全程闭管、无缝衔接,按菌群结构对样本进行快速分类、筛选,减少需要STR分型或二代测序(next-generation sequencing,NGS)的样本数目,降低检验成本,提高检验效率。
3.2 dPCR-HRM 的可行性
近年已有使用dPCR快速分析细菌DNA的报道[31],但均以特定细菌为目标。与单一序列的扩增不同,菌群PCR 不仅需要样本释放足够DNA 模板,还应尽量保持各种细菌DNA 所占的比例。为了验证dPCR 用于菌群分析的可行性,本研究选取5 份唾液样本,每份同时使用dPCR-HRM 和kPCR-HRM 进行菌群分析。两种方法除模板不同外,其他条件保持一致。结果表明,只要两种方法所用模板的样本来源相同,得到的HRM 图谱高度相似,GCP超过80%[GCP>95.85%(96.95%±1.56%)]。这与多数文献[19,22,28]报道的一致,即提取方法对菌群DNA 分析的影响较小。至少对于唾液样本的菌群分型来说,不论直接扩增还是提取后扩增,两者的有效模板构成基本相同,不影响其HRM型的判读。HRM 技术用于菌群分析的可行性已被EVERMAN等[17,32]的研究证实,免于提取的dPCR-HRM在此基础上进一步简化,可在90 min 内同步完成数十甚至上百个样本的分析,同时也更便于操作的自动化。
为了评价dPCR 扩增时菌群模板DNA 释放效率及所需最小样本量,本研究同时用dPCR-HRM 和kPCR-HRM 法调查了一组梯度稀释的唾液样本。该组样本由5 人份唾液等体积混合、梯度稀释而成,以降低个体差异影响,更能代表一般个体的情况。调查结果表明,kPCR-HRM 法可检测低至2.5×10-3ng/μL的唾液DNA,dPCR-HRM 可检测到2.9×10-3nL 的唾液。根据细菌平均基因组大小(约4×106bp)[33]和唾液细菌含量(约9×106/μL)[34]估计,两者大约分别相当于500和20~30 个细菌。虽然试剂盒提取的唾液DNA 中难免混合人和其他微生物的DNA[35],无法准确计算实际的细菌数,但结合两种模板稀释后的唾液量估计,dPCR-HRM 法灵敏度不低于kPCR-HRM 法,支持菌群DNA 可以在本研究的直接扩增条件下有效释放,提取并非必要[28]。
值得注意的是,菌群分析技术不能简单地用传统的灵敏度进行评价。虽然dPCR-HRM 法可检测到稀释3.5×105倍唾液中的细菌,但所得HRM 图谱已与稀释范围在35~3.5×103倍时的明显不同。分析认为,样本稀释倍数过高时,包含的细菌种类已不能代表样本原来的菌群结构,所得HRM 图谱也不具有代表性,且易因随机因素和污染而不稳定。本研究对混检样本的分析结果表明,唾液量在1×10-4~1×10-2μL,相当于1×103~1×105个细菌时,所得HRM 图谱基本一致且稳定。唾液量过高时,可能因干扰物过多或扩增不平衡等,所得图谱也不具有代表性。因此,dPCR-HRM 菌群分析的灵敏度以能获得样本代表性图谱的最小样本量来表示更有实际意义。本研究条件下,dPCR-HRM分析唾液菌群的灵敏度约为1×10-4μL,至少含1×103个以上细菌。
3.3 dPCR-HRM 菌群分析的应用
上述分析结果说明,只有样本用量在一定范围内时,菌群HRM 图谱才有可比性,才能用于分析两个样本是否具有相同或相似的菌群结构。而现场斑痕无法估计沉积体液的量,直接扩增法不能通过DNA 浓度估计细菌数量,必须利用其他信息来判断dPCRHRM 图谱是否能代表样本菌群特征。然而,dPCRHRM 的扩增过程本身具有实时定量的功能。分析上述梯度稀释样本的扩增曲线,结果表明,可以通过样本的Ct 值来估计有效模板量。只要同批检验的两个样本的Ct 值相差不大,即可认为通过HRM 图谱来比较其菌群结构是有意义的。否则,应调整样本用量。实际应用中,应综合分析样本的扩增曲线、熔解曲线以及阴性、阳性对照,结合Ct、GCP 等综合评判。
理想的菌群快速检测还应有较好的鉴别能力、稳定性和检材适应性[36]。为了进一步验证dPCR-HRM鉴别菌群的能力,评价其稳定性和检材适应性,本研究调查了一组唾液样本和唾液斑样本。结果表明,来自不同个体的唾液有不同的HRM 图谱。图谱的个体间差异虽然大小不一[GCP<87.07%(52.11%±22.79%)],但远大于同一样本在不同时间检测时的差异[GCP>92.39%(97.78%±2.30%)]。且同一样本的dPCR-HRM图谱与kPCR-HRM 图谱基本相同,方法间的差异也远小于样本间的差异。虽然样本量有限,但支持dPCRHRM 有较好的鉴别能力和稳定性。
菌群结构的多样性或个体差异是菌群分析法医学应用的主要基础。虽然唾液菌群的高度多样性已被包括NGS 在内的诸多研究[37-38]证实,但dPCR-HRM所能揭示的多样性尚待调查。本研究结果表明,即使受试者均在生活环境和饮食趋同的高校内,仅16S rDNA V4 区就可检出多种类型。且考虑温度、PCR循环次数对HRM 分型的影响,参考SAKARIDIS 等[26]的研究,本研究选择了较为保守的GCP 界值(80%)。未来在体系内引入温度内参、控制循环次数,并在增强稳定性的基础上提高GCP 界值,有望进一步提高dPCR-HRM 技术的分型能力。
检材的复杂性是法医学检验的显著特征,斑痕是最常见的现场检材。本研究对dPCR-HRM 用于唾液斑的可行性进行了初步评估,发现模拟唾液斑用常规的纱线转移法即可进行菌群分析。结合上述灵敏度实验,支持dPCR-HRM 菌群检验并不需要额外的检材处理和消耗,多数情况下常规检材浸洗液即可满足要求。对不同沉积时间唾液斑的调查结果表明,8 h 之内唾液斑与其来源样本的图谱基本相同[GCP>90.83%(96.67%±2.98%)],说明本方法所揭示的斑痕菌群结构是相对稳定的,至少与形成斑痕时的唾液有可比性,这与关于采样时机、采集方法均会影响NGS菌群分析的报道[39]并不一致。结合文献[40]分析认为,与NGS 的高成本、高分辨率不同,本研究所述方法类似于经典的DGGE 菌群分析,分辨率低,对属以下水平变化、低丰度菌属变化和幅度小的变化不敏感。同时,由于唾液含菌量大,高丰度菌属间的比例不易因污染、采样而大幅改变。因此,唾液菌群的HRM 图谱相对稳定,但该方法的分辨率或图谱的个体特异性远小于NGS 分析,其意义主要在于样本初筛,在个体识别方面,可通过HRM 图谱比较快速地排除一部分样本,为重点样本的精准分析创造条件,弥补NGS 成本高、耗时长的局限性。如用于含菌量低、优势菌属不明显的样本,应控制环境、试剂和耗材等可能的细菌污染源,统一采样方式和提取方法,以保证样本间的可比性。
3.4 小结
细菌是法医学检材的重要组成部分。不同体液的菌群有明显差异,同一类型体液的菌群结构也因人而异。现场斑痕、尸体中的菌群包含着丰富的信息,菌群分析是解决一系列法医学问题的新途径。如能实现体液斑痕的菌群快速检测,按体液类型和来源个体对现场斑痕进行分类筛选,有助于明确目标,提高DNA 分型针对性,提高办案效率。因此,研发简单快速的菌群分析技术具有重要意义。
本研究建立了一种不需要提取和电泳的dPCRHRM 菌群分析技术,简单快速且有较高的灵敏度、稳定性和鉴别能力,并初步验证了其在法医学领域的应用。但是,必须意识到菌群分析是以群体特征为目标的微生态学研究,菌群PCR 与传统的单基因座PCR、复合扩增均有诸多区别。本研究仅以16S rDNA V4区和唾液(斑)为例,且只进行了小样本评估,包含温度内参的多片段dPCR-HRM 分析、dPCR-HRM 用于其他类型体液(斑)及应对复杂检材的能力还有待进一步研究。现场斑痕的溯源是法医学的核心问题,虽然人体菌群有高度的个体特异性,但目前还难以通过菌群分析进行个体识别。未来应扩大样本范围和样本量,调查不同采样时机和方法,全面评价各种影响因素,结合菌群溯源技术发展[35,41],积极探索实现法医学斑痕菌群溯源的途径。
