灰黄青霉D-756实时荧光定量PCR体系的建立及验证
2022-03-13严莉洪石烨祺吴君畅瑛施碧红
严莉洪 石烨祺 吴君 畅瑛 施碧红


摘 要:灰黄青霉D-756是1株灰黄霉素高产突变菌株,从转录水平揭示其灰黄霉素合成关键基因的表达情况,有助于了解其基因功能进而对菌株进行定向遗传改造。利用Primer3和Primer-BLAST设计引物,经qPCR验证,引物的扩增产物单一且扩增效率在90%~110%范围内,通过geNorm软件对TUBB、HISH3、RPS24和28S rRNA 4个候选内参基因的表达稳定性进行评价,选择评价结果中较为稳定的候选基因TUBB和RPS24作为内参基因,构建了一个适用于灰黄青霉D-756的qPCR体系。并用该体系检测灰黄青霉D-756中灰黄霉素合成关键基因gsfA在不同培养时间下的相对表达情况,为下一步研究灰黄霉素合成基因簇中各基因功能提供一个有力的工具。
关键词:灰黄青霉;灰黄霉素;荧光定量PCR;内参基因;聚酮合酶
中图分类号:Q 933; Q 78 文献标志码:A 文章编号:0253-2301(2022)01-0006-06
DOI: 10.13651/j.cnki.fjnykj.2022.01.002
Establishment and Validation of the Real-time Fluorescence Quantitative PCR Systemfor Penicillium Griseofulvum D-756
YAN Li-hong, SHI Ye-qi, WU Jun, CHANG Ying, SHI Bi-hong*
(College of Life Science, Fujian Normal University, Fuzhou, Fujian 350108, China)
Abstract: Penicillium griseofulvum D-756 was a mutant strain with high yield of griseofulvin. Revealing the expression of key genes in the biosynthesis of griseofulvin at the transcriptional level would be helpful to understand its gene function and to carry out the directional genetic modification of the strain. The primers were designed by using Primer3 and Primer BLAST. After the qPCR verification, the amplified products of the primers were single and the amplification efficiency was in the range of 90%-110%. Then, the expression stability of the four candidate reference genes TUBB, HISH3, RPS24 and 28S rRNA was evaluated by using the geNorm software. The candidate genes TUBB and RPS24, which were relatively stable in the evaluation results, were selected as the reference genes to construct a qPCR system suitable for Penicillium griseofulvum D-756. Last, this system was used to detect the relative expression of gsfA, the key gene for the biosynthesis of griseofulvin in Penicillium griseofulvum D-756, at different culture times, thus to provide a powerful tool for the further study of each gene function in the biosynthetic gene cluster of griseofulvin.
Key words: Penicillium griseofulvum; Griseofulvin; Fluorescent quantitative PCR; Internal reference gene; Polyketide synthase
灰黃霉素是一种聚酮类抗真菌化合物,主要由青霉属等真菌产生,于1939年由Oxford等分离得到。灰黄霉素能强烈抑制真菌细胞有丝分裂,干扰真菌DNA合成,抑制丝状真菌的生长。它对多种皮肤真菌,如小孢子癣菌属、毛发癣菌属和表皮癣菌属均表现出良好的抗菌活性,并且对人低毒[1],2017年被世界卫生组织纳入基本药物清单,用于口服治疗皮肤癣菌病[2]。在农业上也被用作防治作物真菌性病害,如叶斑病、霜霉病、水稻稻瘟病等[3]。近年来,随着对灰黄霉素抗菌机制的逐步深入研究发现,它对丙型肝炎病毒具有一定的抑制作用[4],且具有潜在的抗癌作用[5]。
20世纪70年代吴松刚等[6]以展青霉Penicillium patulum 4541(系灰黄青霉Penicillium griseofulvum的异名)作为出发菌,利用UV+LiCl等复合诱变处理10多轮获得了灰黄青霉D-756突变株。灰黄青霉D-756菌株具有耐氯且高产灰黄霉素的优良生产特性,发酵液中检测灰黄霉素含量是其出发菌株4541的100多倍,但迄今尚未有对其灰黄霉素高产遗传背景的报道。随着测序技术和实时荧光定量PCR技术的迅速发展,通过分析基因在转录水平的表达量来研究基因功能逐渐成为一种重要的研究手段。
荧光定量PCR技术(qPCR)利用实时荧光信号监测目的基因的扩增,根据Cq值与起始模板量的线性数量关系计算起始模板的表达量,是一种高灵敏度、高通量、强特异性、操作简便的基因定量技术[7]。但正因为其灵敏度高,1个试验参数的变化就会引起试验结果的改变,从而得到不同的试验结论。为了增加试验的可靠性及可重复性,人们制定了一套统一的标准来描述该试验-MIQE(The Minimum Information for Publication of Quantitative Real-Time PCR Experiments guidelines)[8]。利用相对定量方法分析目的基因表达水平的一个关键步骤是利用内参基因对目的基因Cq值进行正确的校正和归一化处理,但目前仅使用单一内参基因作为参照已经很少被接受了,因为几乎很难找到某单个基因可以在不同的培养条件或细胞组织中稳定表达[9]。在丝状真菌中常用的内参基因有TUBB、GAPDH、18S rRNA、28S rRNA、Calmodulin、TefA/Tef1、Actin、HISH3、RPS24等[10-14]。考虑候选内参基因时应选择具有不同功能的内参基因,以降低内参基因之间的共调控作用。
最近本课题组完成了灰黄青霉D-756(P.griseofulvum D-756)的基因组测序,并对灰黄青霉D-756中灰黄霉素合成基因簇进行了分析,发现该基因簇由9个gsf基因构成,包括7个高度保守的gsf基因,即gsfA~gsfF、gsfI,以及2个较不保守的gsf基因(gsfJ、gsfR1)[15]。其中gsfA编码聚酮合酶,负责起始灰黄霉素骨架的合成,是灰黄霉素合成的关键基因。Cacho等[16]通过基因敲除的手段证实了gsfA基因的缺失会阻断灰黄霉素的合成。因此,本研究通过设计荧光定量PCR的引物、筛选稳定表达的内参基因,构建一套适用于灰黄青霉D-756的实时荧光定量PCR体系,并初步探究gsfA基因在不同培养时间下相对表达量的变化,为进一步研究灰黄霉素基因簇各基因功能提供参考依据。
1 料与方法
1.1 试验材料
灰黄青霉D-756菌株。
1.2 试验方法
1.2.1 總RNA提取 分别收集适量培养4、6和10 d的灰黄青霉D-756菌丝样本(每个培养时间设置3个生物学重复,记为4-1、4-2、4-3;6-1、6-2、6-3;10-1、10-2、10-3),经液氮研磨,使用试剂盒提取总RNA(RNA快速提取试剂盒RN40,北京艾德莱),最后进行RNA浓度测定和琼脂糖凝胶电泳检测。
1.2.2 RNA反转录 按照反转录试剂盒(Takara PrimeScriptTM RT reagent Kit with gDNA Eraser)说明书的操作步骤对提取的总RNA进行反转录。首先在gDNA Eraser的作用下42℃ 2 min去除RNA样本中可能含有的基因组DNA,然后将处理过的样本直接进行15 min的反转录反应合成cDNA。
1.2.3 引物设计及合成 选择4个丝状真菌中较为常用的候选内参基因,分别为TUBB、HISH3、RPS24和28S rRNA,连同目的基因gsfA
,以灰黄青霉D-756中各基因的序列作为模板设计引物,经NCBI-ORF Finder找到序列中最保守且不含内含子的区域,利用Primer-BLAST和Primer3(在线版)设计qPCR引物(表1),引物由福州铂尚测序公司进行合成。
1.2.4 引物熔解曲线及标准曲线 将上述设计好的引物按照荧光定量PCR试剂盒(Takara TB Green Premix Ex TaqTM)说明书中的两步扩增法以10 μL反应体系进行qPCR,检测引物的特异性和扩增效率,扩增程序:95℃预变性30 s;95℃ 变性5 s,60℃延伸 30 s,共40个循环;熔解曲线(1个循环):95℃ 10 s,65℃ 1 min,97℃ 1 s;冷却(1个循环):37℃ 30 s。
以50 ng·μL-1 cDNA为起始核酸浓度,按照10倍比稀释形成5个浓度梯度的cDNA样本作为模板,qPCR反应体系TB Green Premix Ex Taq 5 μL,正反引物根据各引物扩增效率添加,ddH2O补足至10 μL,绘制标准曲线,计算扩增效率,选择扩增效率在90%~110%的引物用于后续qPCR试验。
1.2.5 内参基因筛选 利用geNorm软件分析灰黄青霉D-756的4个候选内参基因的Cq值。geNorm是Microsoft Excel中自带的一种应用程式视觉化的Basic脚本(VBA),它可以根据样本Cq值自动计算给定样本中所有内参基因的稳定性M值(M值与基因表达的稳定性成反比),分步排除掉具有最高M值的基因后,剩下的内参基因即是样本中最稳定的内参基因[17]。候选内参基因超过3个时,软件可通过计算内参基因随机组合的M值,进一步得出内参基因的最佳配对数目及组合。
1.2.6 gsfA基因荧光定量表达分析 gsfA基因相对定量分析按照TB Green试剂盒说明书中要求进行qPCR,后采用2-△△Cq法计算gsfA基因在不同培养时间的相对表达量[18]。
2 结果与分析
2.1 总RNA完整性检测
利用1.5%琼脂糖凝胶电泳检测灰黄青霉D-756总RNA的完整性,结果显示3条清晰的条带,分别为28S rRNA、18S rRNA和5S rRNA,其中28S rRNA 条带比18S rRNA条带亮,5S rRNA条带较淡,说明总RNA无降解、完整性较好,可用于下一步实时荧光定量PCR(图1)。
2.2 引物验证
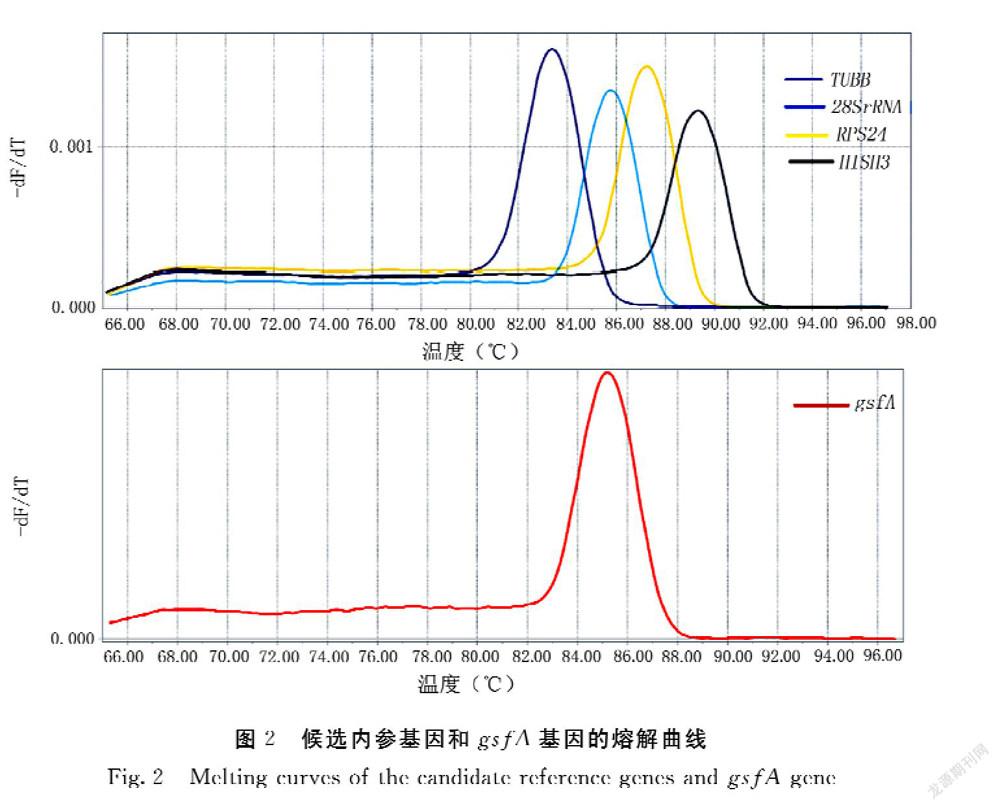
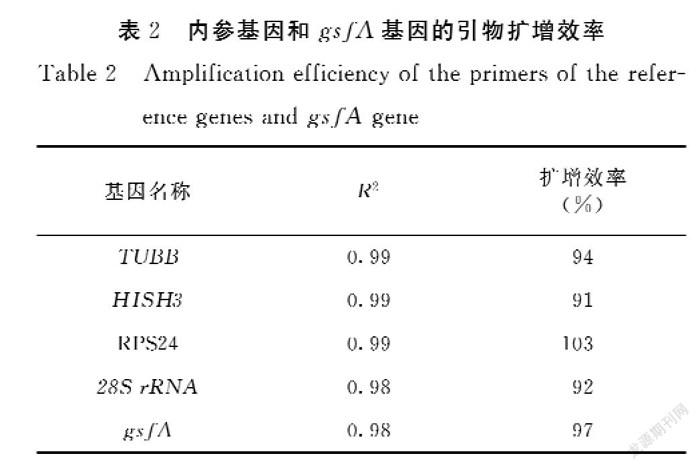
经过qPCR扩增获得引物的熔解曲线,结果显示:4个候选内参基因和gsfA基因的扩增产物的熔解曲线均只出现单一峰,具有良好的特异性,熔解温度在80~90℃(图2)。通过标准曲线进一步确定引物扩增效率,5对引物在0.1~0.3 μmol·L-1浓度范围内的扩增效率为90%~110%,相关系数R2为0.998~1,符合引物的要求(表3)。
2.3 内参基因稳定性分析
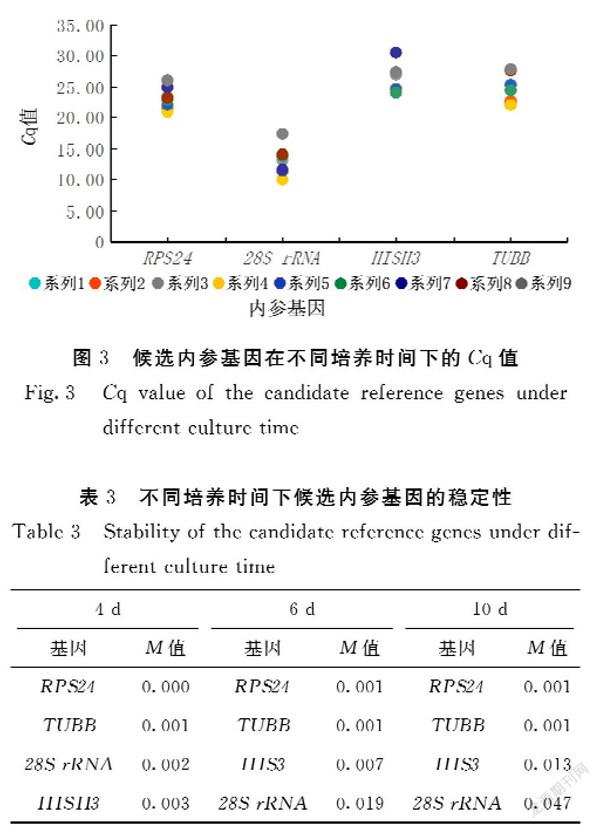
一般来说,理想内参基因的Cq值在15~30,本试验利用qPCR对4个候选内参基因在培养4、6和10 d时的表达量(Cq值)进行分析,结果显示28S rRNA的Cq值低于15,表明其在灰黄青霉D-756中的表达丰度较高,另外3个候选内参基因的Cq值均在合理范围内(图3),可用作下一步内参基因稳定性分析。
经geNorm软件分析4个候选内参基因在培养4、6和10 d时的表达量(Cq值),获得稳定性M值(表3),以稳定性M值=1.5为界限,该数值越接近0说明该内参基因的表达越稳定。结果显示在不同培养时间里4个内参基因的稳定性M值均小于1.5,表明候选内参基因的表达都较稳定。由表3可知,在不同培养时间下,M值最小的均为RPS24,说明经geNorm软件分析
RPS24的稳定性最好。在培养4 d时,HISH3的M值最大;在培养6、10 d时,28S rRNA的M值最大,说明HISH3和28S rRNA的稳定性较差。利用geNorm软件进一步分析候选内参基因的最佳配对数目及组合,结果表明选择RPS24与TUBB
两个基因作为内参基因组合可获得最佳的稳定性,M值为0.011。因此,本试验从4个候选内参基因中筛选得到2个内参基因,即RPS24与TUBB组合,后续以二者Cq值的几何平均值作为内参基因的Cq值计算灰黄青霉D-756中gsfA基因的相对表达量。
2.4 gsfA基因表达量分析
根据geNorm软件分析得出的结果,选择RPS24与TUBB组合作为内参基因,检测灰黄青霉D-756中gsfA基因在摇瓶培养4、6和10 d的相对表达量。单因素方差分析结果表明,总体上gsfA基因在不同培养时间(4、6、10 d)的相对表达量无显著差异(F=3.688073,DF=2,P=0.090),其中第4 d与第6 d的相对表达量无显著差异(T=-1.388, DF=4,P=0.237),第6 d与第10 d的相对表达量差异较大(T=2.357,DF=4,P=0.078)。从趋势上看,gsfA在第4 d到第6 d的表达水平逐渐升高,并在第6 d时达到最高,平均相对表达量为306.60,而在第6 d到第10 d的表达水平逐渐下降,10 d时平均相对表达量为35.53。
3 结论与讨论
本试验选择了4个候选内参基因(TUBB、HISH3、RPS24和28S rRNA),除28S rRNA的Cq值小于15之外,其余3个候选内参基因的Cq值都在合理范围。利用geNorm软件对4个候选内参基因在灰黄青霉D-756不同培养时间(4、6和10 d)下的表达量进行分析,结果表明单个基因表达最稳定的是RPS24,TUBB次之,28S rRNA和HISH3的稳定性在4个候选内参基因中较差。而将RPS24与TUBB组合作为内参基因时,它们的稳定性更佳,因此最终选择RPS24与TUBB组合作为灰黄青霉D-756的内参基因用于目的基因表达量的标准归一化。
引物设计对于实时荧光定量PCR方法来说也是十分重要的。本试验首先利用NCBI-ORF Finder预测序列中最保守的外显子序列,以此为模板利用Primer-BLAST和Primer3(在线版)设计qPCR引物。经qPCR的熔解曲线仅出现一个单峰,表明利用该方法设计的引物特异性良好。进一步通过设置5个浓度梯度的cDNA样本作为模板以及在0.1~0.3 μmol·L-1范围内调整引物浓度制作标准曲线检测引物的扩增效率,找到了不同引物的最佳浓度,使得扩增效率在90%~110%区间内,从而为qPCR的定量提供准确性。
最后,将构建的qPCR体系用于检测灰黄青霉D-756的gsfA基因在本试验条件下培养4、6和10 d的表达情况。根据CHOOI等[19]阐述的灰黄霉素生物合成途径,聚酮合酶GsfA起始灰黄霉素的合成,即一个乙酰辅酶A和六个丙二酸单酰辅酶A起始单元在gsfA基因编码的非还原聚酮合酶作用下生成灰黄霉素骨架-二苯甲酮,之后在灰黄霉素合成基因簇中其他酶的作用下生成终产物灰黄霉素。在灰黄青霉D-756菌株中,gsfA基因的表达情况与其近缘菌株灰黄青霉PG3的结果相似[14],在整个培养期间(4~10 d),gsfA基因的相对表达量无显著性差异。培养4~6 d时,D-756菌株gsfA基因的相对表达量保持较高水平(合成骨架需要),培養10 d时,D-756菌株中gsfA基因的表达水平出现下降,这与gsfA基因在灰黄霉素合成过程中的作用基本一致。本试验为后续开展灰黄青霉D-756灰黄霉素生物合成基因簇各基因表达的分析打下了基础。
参考文献:
[1]HAMDY A K, SHEHA M M, ABDEL-HAFEZ A A, et al.Design, Synthesis, and Cytotoxicity Evaluation of Novel Griseofulvin Analogues with Improved Water Solubility[J].International Journal of Medicinal Chemistry,2017,2017:7386125.DOI:10.1155/2017/7386125.
[2]WHO Expert Committee on Selection, Use of Essential Medicines, World Health Organization.The Selection and Use of Essential Medicines: Report of the WHO Expert Committee, 2017(including the 20th WHO Model List of Essential Medicines and the 6th Model List of Essential Medicines for Children)[M].Geneva,World Health Organization, 2017.
[3]张传能,黄铭杰,毛宁.灰黄霉素对水稻稻瘟病菌的防治效果研究[J].中国农学通报, 2015, 31(4):190-194.
[4]JIN H, YAMASHITA A, MAEKAWA S, et al.Griseofulvin, an oral antifungal agent, suppresses hepatitis C virus replication in vitro[J].Hepatol Res,2008, 38(9): 909-918.
[5]REBACZ B, LARSEN T O, CLAUSEN M H, et al.Identification of griseofulvin as an inhibitor of centrosomal clustering in a phenotype-based screen[J].Cancer Research, 2007, 67(13): 6342-6350.
[6]吴松刚,郝家骥,戚晓玉,等.Penicillum patulum 4541耐前体突变株的选育[J].遗传, 1980, 2(4): 16-18.
[7]BUSTIN S A, BENES V, NOLAN T, et al.Quantitative real-time RT-PCR-a perspective[J].J Molecular Endocrinology, 2005, 34(3): 597-601.
[8]BUSTIN S A, BENES V, GARSON J A, et al.The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments[J].Cliniacl Chemistry, 2009, 55(4): 611-622.
[9]VANDESOMPELE J.Reference Gene Validation Software for Improved Normalization[J].Real-time PCR: current technology and applications, 2009, (4): 47-64.
[10]BARKER B K K, VDISCH M, MAZURIE A, et al.Transcriptomic and proteomic analyses of the Aspergillus fumigatus hypoxia response using an oxygen-controlled fermenter[J].BMC Genomics, 2012, (13): 62.DOI: 10.1186/1471-2164-13-62.
[11]ARCHER M, XU J.Current Practices for Reference Gene Selection in RT-qPCR of Aspergillus: Outlook and Recommendations for the Future[J].Genes (Basel), 2021, 12(7): 960.DOI: 10.3390/genes12070960.
[12]ZAMPIERI D, NORA L C, BASSO V, et al.Validation of reference genes in Penicillium echinulatum to enable gene expression study using real-time quantitative RT-PCR [J].Current Genetics, 2014, 60(3): 231-236.
[13]LEVIN E, RAPHAEL G, MA J, et al.Identification and Functional Analysis of NLP-Encoding Genes from the Postharvest Pathogen Penicillium expansum[J].Microorganisms, 2019, 7(6): 175.DOI: 10.3390/microorganisms7060175.
[14]BANANI H, MARCET-HOUBEN M, BALLESTER A R, et al.Genome sequencing and secondary metabolism of the postharvest pathogen Penicillium griseofulvum[J].BMC Genomics, 2016, (17): 19.DOI: 10.1186/s12864-015-2347-x.
[15]PARISA A, LIHONG Y, YULONG W,et al.Conservation of griseofulvin genes in the gsf gene cluster among fungal genomes[J].G3(Genes Genomes Genetics), 2021.jkab399.DOI:10.1093/g3journal/jkab399.
[16]CACHO R A, CHOOI Y H, ZHOU H, et al.Complexity generation in fungal polyketide biosynthesis: a spirocycle-forming P450 in the concise pathway to the antifungal drug griseofulvin[J].ACS Chemical Biology, 2013, 8(10): 2322-2330.
[17]VANDESOMPELE J, DE PRETER K, PATTYN F, et al.Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes[J].Genome Biology,2002, 3(7): RESEARCH0034.DOI: 10.1186/gb-2002-3-7-research0034.
[18]LIVAK K J, SCHMITTGEN T D.Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta DeltaC(T)) Method[J].Methods, 2001, 25(4):402-408.
[19]CHOOI Y H, CACHO R, TANG Y.Identification of the viridicatumtoxin and griseofulvin gene clusters from Penicillium aethiopicum[J].Chemical Biology, 2010, 17(5):483-494.
(責任编辑:柯文辉)
收稿日期:2021-12-10
作者简介:严莉洪,女,1995年生,硕士研究生,主要从事真菌分子遗传学研究。
通信作者:施碧红,女,1968年生,博士,教授,主要从事微生物生化与分子生物学等研究(E-mail:shibh@fjnu.edu.cn)。
基金项目:国家自然科学基金(91435106);2022年福建师范大学生命科学学院“溪源江学者”科研创新项目(22FSSK015)。
3044501908206
