High-throughput computational material screening of the cycloalkane-based two-dimensional Dion-Jacobson halide perovskites for optoelectronics
2022-03-12GuoqiZhao赵国琪JiahaoXie颉家豪KunZhou周琨BangyuXing邢邦昱XinjiangWang王新江FuyuTian田伏钰XinHe贺欣andLijunZhang张立军
Guoqi Zhao(赵国琪), Jiahao Xie(颉家豪), Kun Zhou(周琨), Bangyu Xing(邢邦昱),Xinjiang Wang(王新江), Fuyu Tian(田伏钰), Xin He(贺欣), and Lijun Zhang(张立军)
State Key Laboratory of Integrated Optoelectronics,Key Laboratory of Automobile Materials of MOE,College of Materials Science and Engineering,Jilin University,Changchun 130012,China
Keywords: first-principle calculations, two-dimensional halide perovskites, electronic structures, Dion-Jacobson phaseperovskites,optoelectronic applications
1. Introduction
Conventional three-dimensional (3D) organic-inorganic halide perovskites (OIHP) have recently undergone unprecedented rapid development.[1,2]They possess outstanding optical ad electronic properties such as strong light absorption, excellent charge transportation,[3]high defect tolerance along with solution processability.[4]The power conversion efficiency of perovskite solar cells has remarkably increased from approximately 3% to greater than 25%.[5]However,their inherent environmental instability under moisture, light, and heat remains crucial hurdles prior to the realization of commercialization.[6]There poor environmental resistance primarily results from their susceptibility to light-induced ionic diffusion and the volatility of the organic cations. In contrast to the volatile small cations such as methylammonium (MA+) and formamidinium (FA+) that are used in traditional 3D perovskites, the bulky organic cations incorporated within two-dimensional(2D)perovskites exhibit greater environmental resistance.[7-9]Dion-Jacobson(DJ) phases 2D halide perovskites attract significant research interest due to their long-term stability under ambient conditions,significant moisture resistance,rich chemical and structural diversity, and consequently widely tunable electronic properties.[10,11]The DJ-phases 2D perovskite has the general formulaABn-1MnX3n+1,whereAis a divalent organic cation,Bis a monovalent cation(such as methylammonium MA+and formamidinium FA+),Mis a divalent metal,Xis a halide anion, andnis defined as the layer number of corner-sharing[PbI6]4-inorganic slabs.[12,13]The strong chemical bonding between organic spacing cations and inorganic frameworks has been demonstrated to enhance the structural stability of DJ perovskites.[7,10]Nevertheless, the DJ-phases perovskites are normally not excellent candidates as light-absorbers for photovoltaic applications in view of their broad optical bandgap[9]and limited charge-carrier transportation in the out-of-plane direction within the 2D structure.[14]
In recent years, many efforts have been made to improve the photovoltaic properties of DJ-phase perovskites by designing theA-site organic spacing cation.[15]The organic spacing cation influences the distortion of the inorganic layers, which further impacts on the optical and electronic properties.[3,16]The first DJ perovskite solar cell was reported by using 3-(aminomethyl)piperidinium(3AMP)and 4-(aminomethyl)piperidinium(4AMP)as the organic spacing cations.[17]The best power conversion efficiency is 7.32%for the form (3AMP)(MA)3Pb4I13(n= 4).[18,19]Compared to the aliphatic cation of the same size, the aromatic spacers increase the rigidity of the cation, reduce the interlayer spacing, and decrease the dielectric mismatch between the inorganic layer and the organic spacer. Kanatzidiset al.have reported the (3AMPY)(MA)3Pb4I13-based devices ((3AMPY=3-(aminomethyl)pyridinium)) which has a champion power conversion efficiency of 9.20%.[10]Later on, the short divalent ligand propane-1, 3-diammoniym(PAD) based DJ-phase perovskite solar cell with 13.3% efficiency for (PAD)(MA)3Pb4I13(n=4) was reported.[20]Until now, based on straight-chain diammonium cation 1, 4-butanediamine(BDA),Zhenget al. reported a power conversion efficiency of 16.38% for (BDA)MA4Pb5I16(n=5).[20]Wanget al.have obtained 15.01% power conversion efficiency for (CHDA)MA3Pb4I13(n= 4, CHDA=trans-1, 4-cyclohexanediamine) DJ-phase perovskite, and the unsealed devices retain 94.7%of their initial efficiency for CHDA based devices under the 1248 h storage inside 50%-60%relative humidity dark cabinet.[21]While these materials show competitive photovoltaic performances, the underlying mechanism is worth exploring. Based on this situation, we study and compare a series of the cycloalkane organic molecule based 2D DJ perovskites.[22]
Compared to unsaturated lipids, cycloalkane (CA) organic molecules possess better chain flexibility and folding properties, which may improve the stability of the DJ perovskite.[23]In this work, a serious of cycloalkane molecules were selected as the organic spacing cation in 2D DJ perovskites, which can widely manipulate the optoelectronic properties of the DJ perovskites.[10]We investigated how the structure of CA molecular spacer would influence the crystallinity behavior,and further impact on the thermodynamic, electronic and optoelectronic properties by using automatic high-throughput workflow cooperated with densityfunctional(DFT)calculations.[24,25]Before the cosmically research,the influence of the thickness of the inorganic layer on the thermal dynamical stability was examined.[25]Based on the tests,we used 4 inorganic octahedron layers extracted from the (001) direction of methylamine lead iodide to construct the CA-based DJ perovskite.[26]Three types of diaminocycloalkane molecules classified by the ways of amino linkage have been considered. Each type includes 9 or 10 molecules with carbon numbers ranging. In addition,the molecule orientations of cis and trans configurations are considered. In total 58 CA-based DJ halide perovskites have been studied. We found that the CA-based DJ perovskites are all thermal dynamically stable. The detailed structural analysis presents the deformations of the inorganic layers, CA molecules and the whole lattice. Combined with electronic structure properties,we found the most proper CA-based DJ perovskites for light absorber materials. At the end,we also analyzed the quantum well structure of all designed CA-based DJ perovskites. The results presented in this work fill in the research blank in CAbased DJ perovskites and contribute to a more thorough understanding of the structural-properties relationship in OIHPs.
2. Computational methods
The first-principles calculations presented in this paper were performed under the framework of density-functional theory (DFT) using the projector augmented wave (PAW)potential method[24]implemented in the Viennaab initiosimulation package.[26]The generalized gradient approximation(GGA)proposed by Perdew-Burke-Ernzerhof(PBE)[27]was selected as the exchange-correlation functional. The PAW pseudopotentials with 2s22p2(C), 2s22p3(N), 1s1(H),5d106s26p2(Pb), and 5s25p5(I) treated explicitly as valence electrons were used to describe the electron-ion interactions.AΓ-centeredk-point mesh of 1×5×5 and a plane-wave energy cutoff of 500 eV were adopted for electronic Brillouin zone integration. All lattice constants and atomic positions were optimized through total energy minimization until the residual forces on atoms dropped below 0.01 eV/°A. The energy converge criterion of 10-5eV was used in all charge distribution optimization calculations. To describe the longrange van der Waals (vdWs) interaction which plays an important role in organic-inorganic halide perovskite materials, the optb86b-vdW functional[28]was used in all calculations. The structure constructions,calculations and data analysis were performed by the automatic high-throughput materials research workflow in the Jililn Artificial-intelligence aided Materials-design Intergraded Package (JAMIP),[29]which greatly accelerated the standard research process. The carriers’ effective mass calculations were based on the semiclassical Boltzmann transport theory[24]that takes into account effects of non-parabolicity and anisotropy of bands, multiple minima,multiple bands,etc. on the carrier transport.
It is well known that standard GGA-PBE methods underestimate the band gaps, hence we recalculated the band gap (Eg) with the Heyd-Scuseria-Ernzerhof (HSE06) hybrid functional approach. The spin-orbit coupling (SOC) was taken into consideration due to the non-negligible effect on the heavy metals, especially Pb. We have chosen the cis and trans structures of C4 based DJ halide perovskites for each cycloalkane molecule type and recalculated the band gap of these 6 perovskites by using HSE+SOC.The results are shown in Table 1. The band gap of each constructed DJ perovskite was then corrected by a scissor operator equal to the band gap difference between the HSE+SOC and PBE calculations for the C4 based DJ halide perovskite constructed with the same type and orientation of the cycloalkane. For the carriers’ effective mass, a high-densityk-point grid is required in their calculations, making it too expensive for them to apply the HSE+SOC method. Such inspection can be carried out in the future works of these compounds.
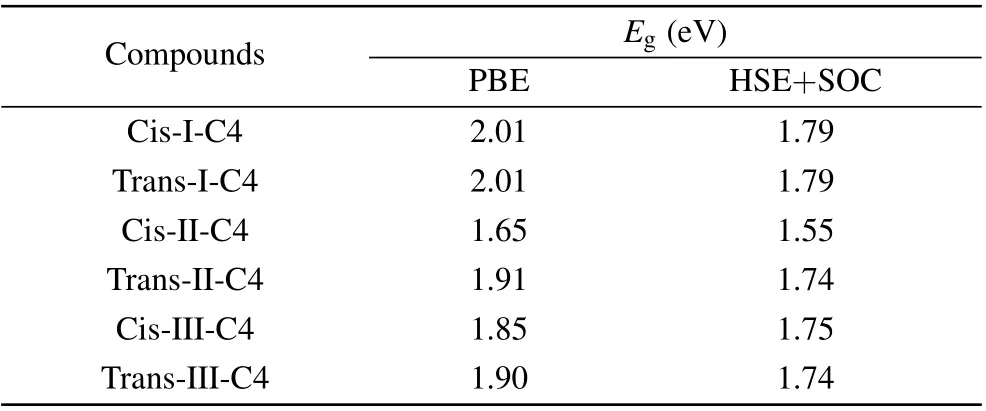
Table 1. The calculated band gap (Eg) by using PBE and HSE+SOC methods and carrier effective mass(m*e for electron and m*h for hole)of the cis and trans structures of C4 based DJ halide perovskites for each cycloalkane molecule type by using PBE method with and without considering the SOC effect.
3. Results and discussion
3.1. Crystal structures and thermodynamic stabilities
In this work, a series of cycloalkane organic molecules were selected as theA-site organic spacing cation in 2DDJ halide perovskite. The flexible property of cycloalkane organic molecules makes them suitable as the interlayer spacers for 2DDJ perovskites. Since this research focuses on organic hybrid perovskites with DJ phases, the cycloalkanes with two NH+3substituents are considered. The formula is [(CmH2m-2)(NH3)2](MA)n-1PbnI3n+1(n= 4,m=3,4,...,12), wherenis the layer number of corner-sharing[PbI6]4-inorganic slabs andmis the number of carbon atoms in the cycloalkane ring. In total, 29 naphthenic molecules containing double functional groups of NH+3were selected as spacer cations. These molecules have been synthesized in the experiment and the structure diagrams are shown in Fig.1(a). According to the relative position of the NH+3substitution within the carbon ring, we divided these diaminocycloalkane molecules into three categories: type I: two NH+3groups are connected to one carbon atom in the carbon ring;type II:two NH+3groups are connected to two adjacent carbon atoms in carbon rings; type III: NH+3groups are connected to the spaced carbon atoms in carbon rings. These diaminocycloalkane molecules are named by the number of carbon atoms in the ring for each type(C3,C4,...,C12).
In 2D perovskites,the number of corner-sharing[PbI6]4-inorganic layer can influence the thermodynamic stability and electronic properties. We have tested the number of inorganic layers in 2D DJ perovskites based on thermodynamic stability. In order to evaluate the stability of the 2D perovskites, we calculated the formation energy(ΔH) defined as the energy difference between the original perovskite and decomposition products. Consider the reaction pathway [(CmH2m-2)(NH3)2](MA)n-1PbnI3n+1→(CmH2m-2)(NH3)2I2+(n-1)(MA)I+(3n+1)PbI2, the formation energy ΔHof the CA-based DJ halide perovskites[(CmH2m-2)(NH3)2](MA)n-1PbnI3n+1was calculated using the following formula:
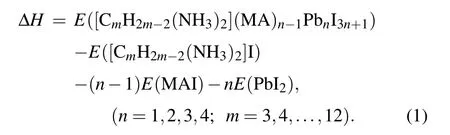
We used C10 diaminocycloalkane molecules for each type as the interlayer spacer to construct the crystal structures of DJ perovskites with different inorganic layers (n= 1 to 4). The calculated results are shown in Fig. 1(d). The negative value of ΔHcorresponds to a thermodynamically stable condition. All these structures have negative ΔH, which indicates that these structures are thermodynamically stable.The more stable the material, the large negative magnitude of the formation energy. In comparison, the four inorganic layers based DJ perovskite has the most stable structure. In this work, we focus on the [(CmH2m-2)(NH3)2](MA)3Pb4I13(n= 4,m= 3,4,...,12) DJ perovskites. Then, we investigated the effect of the orientation of diaminocycloalkane molecules on DJ perovskites. We considered the relative orientation of the two diaminocycloalkane molecules in the unit crystal cell. The dipole of the diaminocycloalkane molecules is in the same orientation (cis) or in the opposite orientation (trans) in the unit crystal cell. The crystal structures of cis-[(C10H18)(NH3)2](MA)3Pb4I13and trans-[(C10H18)(NH3)2](MA)3Pb4I13DJ perovskites are shown as an example in Figs. 1(b) and 1(c). Finally, 58 diaminocycloalkane based DJ halide perovskites have been investigated in this work.
We turn our attention to the thermodynamic stability of these DJ perovskites. Figure 1(e) presents the calculated results of ΔH.[28]The formation energies of these DJ perovskites are all negative,which indicates that they are thermodynamic stable. With the increase of the number of carbon atoms in the interlayer molecules, the formation energy tends to increase, which indicates that the stability of the DJ perovskite decreases. The increased size of the interlayer molecule leads to the distortion of the inorganic framework, this may be the reason for the decreased stability. In addition, the type III interlayer molecular based DJ perovskite is more stable than type II and type I based ones, no matter in the cis or trans orientation. Type III diaminocycloalkane molecules are more suitable as the interlayer spacers for the DJ perovskite.
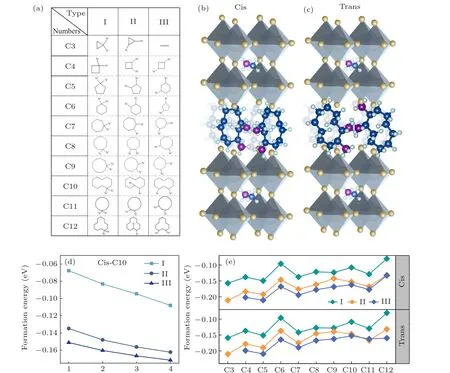
Fig. 1. (a) The structure diagram of 29 diaminocycloalkane molecules, which we considered as the interlayer spacer in two-dimensional (2D)Dion-Jacobson (DJ) phases halide perovskites. According to the relative position of the NH+3 substitution within the carbon ring, these diaminocycloalkane molecules have been divided into three categories: type I: two NH+3 groups are connected to one carbon atom in the carbon ring; type II: two NH+3 groups are connected to two adjacent carbon atoms in carbon rings; type III: NH+3 groups are connected to the spaced carbon atoms in carbon rings. These diaminocycloalkane molecules are named by the number of carbon atoms in the ring for each type (C3,C4, ..., C12). The crystal structure of our inorganic layered DJ perovskites based on C10 diamino cycloalkane cation with the formula (b) cis-[(C10H18)(NH3)2](MA)3Pb4I13 and (c) trans-[(C10H18)(NH3)2](MA)3Pb4I13. The dipole of diamino cycloalkane molecule C10 is in the same orientation in cis-[(C10H18)(NH3)2](MA)3Pb4I13 and in the opposite orientation in trans-[(C10H18)(NH3)2](MA)3Pb4I13. (d)The calculated formation energy(ΔH)of[(C10H18)(NH3)2](MA)n-1PbnI3n+1 (n=1,2,3,4). The C10 diamino cycloalkane molecules for each type are used as the interlayer spacer in different inorganic layers(n=1-4)of 2D DJ perovskites. (e)The calculated ΔH of 58 DJ perovskites with formula cis-and trans-[(CmH2m-2)(NH3)2](MA)3Pb4I13 (n=4,m=3,4,...,12).
The organic spacing cation influences the distortion of the inorganic layers, which has a further impact on the thermodynamic stability and electronic properties. In order to gain a deeper understanding of the relation between diaminocycloalkane interlayer molecules and crystal structures, we investigated the structural distortion of the inorganic framework.Here, we used the average Pb-I-Pb bond angle between the PbI6octahedron, the average Pb-I bond length, the I-Pb-I bond angle variance(δ2)in the PbI6octahedron and the interlayer distance to evaluate the distortion degree of the inorganic framework. The formula ofδ2is shown below:
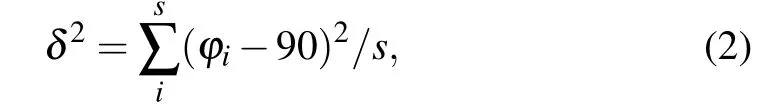
whereφiis the I-Pb-I bond angles for the octahedra,smeans the number of I-Pb-I bond angle.In an ideal PbI6octahedron,the I-Pb-I bond angle is equal to 90°,σ2is equal to 0. Largerδ2indicates higher octahedra distortion.The results are shown in Figs.2(a)-2(d).
In an ideal cubic perovskite with no octahedral tilting,the Pb-I-Pb bond angle is equal to 180°. The distortion degree of the adjacent PbI6octahedron is expressed by the degree of the average Pb-I-Pb bond angle deviating from 180°. From Fig. 2(a), with the number of carbon atoms in the interlayer molecule increasing,the degree of average Pb-I-Pb bond angle deviating from 180°is decreasing. The distortion degree between the adjacent PbI6octahedron is decreasing with the size of diamino cycloalkane molecule increasing. The relationship between the average length of the Pb-I bonds in the PbI6octahedron and the number of carbon atom is presented in Fig. 2(b). The I atoms which are far away from the interlayer spacer in the inorganic octahedron are little affected by the organic molecule, thus the average bond length does not change significantly. Meanwhile, the error bar in Fig. 2(b)is calculated based on the variance in the length of the Pb-I bond,which represents the range of Pb-I bonds. The range of the error bar also increases with the increase in the number of carbon atoms, which indicates the distortion of the inorganic framework.
The value of I-Pb-I bond angle variance(δ2)can evaluate the distortion degree of PbI6octahedron. As we can see in Fig. 2(c),δ2is impacted by the relative orientation of the interlayer molecules. The trans structures are more sensitive to the size of the interlayer molecule than the cis structures.The larger the interlayer molecule,the larger distortion degree of PbI6octahedron. The interlayer distance of these DJ perovskites also has the same trend (Fig. 2(d)). The distance of neighbor Pb atom in inorganic layer and N atom in interlayer spacer is also analyzed to investigate the interaction between the inorganic framework and interlayer spacer. The distance between inorganic framework and interlayer spacer becomes larger as the size of the interlayer molecule increases.
Furthermore, we consider the change in diamino cycloalkane interlayer molecule.We focus on the change of C-N bonds between the carbon ring and the NH3functional group.The results are shown in Fig. 2(f). The normal length range of the C-N bond is 1.47 °A to 1.50 °A.The average C-N bond lengths larger than 1.50 °A are located in the dark blue shade region. Obviously,when the number of carbon atoms is larger than C7, the C-N bond length is greater than 1.50 °A, which means that the diamino cycloalkane interlayer molecules are stretched. This might be indicating that the interaction force between the inorganic framework and interlayer molecule influences the bond length of the NH3functional groups.
In summary, as the size of the interlayer molecule increases, it further increases the distortion degree of the inorganic framework of the DJ perovskites. This is responsible for the thermodynamic stability decrease.
Fig. 2. The (a) average Pb-I-Pb bond angle, (b) average Pb-I bond length, (c) I-Pb-I bond angle variance, (d) interlayer distance, (e) distance of neighbor Pb atom in inorganic layer and N atom in interlayer spacer,and(f)average C-N bond length in the diamino cycloalkane interlayer molecule vs. the number of carbon atom in the diamino cycloalkane molecule in DJ perovskites. The region of average C-N bond length larger than 1.5 °A is marked by the dark bule shade,which means that the diamino cycloalkane interlayer molecules are stretched.
3.2. Electronic structure
3.2.1. Bandgap and effective mass distribution
Typical organic-inorganic halide perovskites such as MAPbI3have an average bandgap of about 1.5 eV and belong to the semiconductors which have a wide absorption range.With the introduce of big organic spacers, the structural connectivity and deformations of the original perovskites structure are substantially changed, thus making a great influence on the electronic properties. As discussed in Section 2,the cycloalkane molecules can effectively tune the deformations in the perovskite structure,which enables the tuning of the electronic structures as well.
As shown in Fig. 3(a), the calculated bandgaps of the cycloalkane-2DOIHP are in the range of 0.87-1.93 eV,which is a very wide tuning window. Especially in cis-oriented type 1 and type 2 cycloalkane-2DOIHPs,the wide distributed bandgaps(0.68-1.93 eV)show a quasi-linear decreasing trend with increasing cycloalkane carbon numbers. This trend can be interpreted by the structural deformations introduced by the cycloalkane molecules. With increasing molecule size,the interlayer distances will increase, but the C-N bonds are also elongated, indicating an increasing interaction between the molecules and inorganic layer. However, cycloalkane-2DOIHPs with trans-oriented cycloalkane do not possess wide distributed bandgaps(1.51-2.1 eV).This could be attributed to the general large distortions induced by the trans-oriented cycloalkane,which break the bandgap decreasing trend appeared in the cis-oriented cycloalkane-2DOIHP.
The carrier effective masses are also calculated. As shown in Fig.3(b), the cycloalkane-2DOIHPs generally have outstanding charge transport properties,with electron and hole effective mass in the range of 0-1m0(m0is the free electron mass), except for some highly distorted structures. Suitable optical band gap is a primary prerequisite for specific optoelectronic applications. Combine with the small carrier effective mass[30](m*eorm*h<m0),which is benefit for the carrier transport properties. We screened the band gaps in range 1.0-1.7 eV, which are optimum for photovoltaic materials, e.g.,cis-I-C7 and cis-II-C10. The cis-III-C8 compound has a direct band gap of 1.67 eV,which might be suitable for red light LED application. Further experimental research on these 2D perovskites is called for verifying our theoretical predictions.
Fig.3. Bandgaps and carrier effective mass distribution of cycloalkane-based 2DDJs. (a)The bandgap values of all cycloalkane-based 2DDJs are scissors-shifted to match the HSE+SOC gap. Each type of molecule is labeled out with dots of different colors. The direct bandgap and indirect bandgap are marked with solid and hollow dots,respectively. (f)The carriers effective mass of all cycloalkane-2DDJs. Hole and electron effective masses are marked with solid and hollow dots,respectively.

Fig.4. The projected band structures of cis oriented type 2 C3, C8, C10 and C12 molecules cycloalkane-2DDJ.The Pb-p, Pb-s, I-p orbitals and contributions from cycloalkane2+ and CH3NH+3 are presented with orange, red, purple and blue dots, respectively. The band gap of the band structure is scissors-shifted to match the HSE+SOC gap.
3.2.2. Band structure and density of states
To better understand the electronic properties,now we examine the band structures and corresponding density of states(DOS) of four selected cycloalkane-2DOIHP: cis-oriented typ 2 C3, C8, C10 and C12. We project the band structure and DOS to five components: p orbitals of Pb atoms, s orbitals of Pb atoms, p orbitals of I atoms, all CH3NH+3atoms and all cycloalkane2+atoms,represented by orange,red,purple and blue colors, respectively. From Figs. 4(a) and 5(a),we can see that the band edges are composed of I-p, Pb-s orbitals for VBM and Pb-p,I-p orbitals for CBM,which are the same with methylamine lead iodide. These band edges compositions also explain the origin of the good charge transport properties. The energy level of the cycloalkane molecule is generally away from the band edge (Figs. 4(a) and 5(a)), but with the increasing size of the cycloalkane molecule, its energy level also increases and becomes more approaching to the valence band edge (Figs. 4(b)-4(d) and 5(b)-5(d)). This energy level shift pushes up the valence band and reduces the band edge dispersion, which is most pronounced in the C12 cycloalkane-2DDJ(Fig.4(d)). However,this valence band energy level shift does not directly determine the bandgap. As we can see from Fig.4,the bandgap change is mostly induced by the descending of the conduction band.With increasing cycloalkane size (C3-C8-C10), the conduction band gets more dispersive, and the bandgaps are getting smaller. In C12, the conduction band is more dispersive than that in C3, but less dispersive than that in C8 and C10, thus leading a bandgap between these two materials. This can be understood by considering the overlarge inorganic layer deformation induced by the oversized cycloalkane. The too large deformation pushes up the conduction band again.
Fig.5. The projected density of states of cis oriented type 2 C3,C8,C10 and C12 molecules cycloalkane-2DDJ.The Pb-p,Pb-s,I-p orbitals and contributions from cycloalkane2+ and CH3NH+3 are presented with orange,red,purple and blue lines,respectively.
4. Conclusion and perspectives
To explore the properties of cycloalkane-based Dion-Jacobson (DJ) halide perovskites, we constructed a detailed chemical space containing three types of cycloalkane (CA)molecules labeled by the ways of amino linkage. Each cycloalkane molecule type includes 9 or 10 molecules with carbon numbers ranging from 3 to 12. The relative orientation of the two cycloalkane molecule configurations was considered as well.High-throughput calculations revealed that all the CAbased DJ halide perovskites in the chemical space were thermal dynamically stable. By detailed analysis of structural lattice deformations and electronic structure properties,we found that the cycloalkanes can effectively tune the structural deformations and electronic properties including bandgaps and effective mass and realized a wide bandgap tuning range of 0.9-2.1 eV.Finally,we found that cycloalkanes with carbon atoms number of 5-8 will lead to a relatively small deformation in the lattice and a more suitable bandgap and carriers’effective mass for light absorb applications.
Data availability The data that support the findings of this study are available in Science Data Bank, with link http://www.doi.org/10.11922/sciencedb.j00113.00006.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 62004080), the Postdoctoral Innovative Talents Supporting Program (Grant No.BX20190143),and the China Postdoctoral Science Foundation (Grant No. 2020M670834). Calculations were performed in part at the high-performance computing center of Jilin University.
猜你喜欢
杂志排行

Chinese Physics B的其它文章
- Surface modulation of halide perovskite films for efficient and stable solar cells
- Graphene-based heterojunction for enhanced photodetectors
- Lithium ion batteries cathode material: V2O5
- A review on 3d transition metal dilute magnetic REIn3 intermetallic compounds
- Charge transfer modification of inverted planar perovskite solar cells by NiOx/Sr:NiOx bilayer hole transport layer
- A low-cost invasive microwave ablation antenna with a directional heating pattern