副猪嗜血杆菌Flp-FRT双基因敲除方法的建立与优化
2022-03-08肖坤雪王翘楚陈焕春蔡旭旺徐晓娟
肖 静,肖坤雪,王翘楚,陈焕春,蔡旭旺,徐晓娟
(华中农业大学动物医学院 农业微生物学国家重点实验室,湖北省预防兽医学重点实验室,武汉 430070)
副猪嗜血杆菌(Glaesserellaparasuis,G.parasuis)是存在于猪上呼吸道的条件致病菌,可引起以纤维性多发性浆膜炎、关节炎、脑膜炎、急性肺炎和败血病为特征的猪格氏病(Glässer’s disease),许多生猪养殖国家受其危害[1-3]。但目前对G.parasuis毒力因子和致病机制的了解还不够充分,给猪格氏病的防控和疫苗研发带来困难[4-5]。其中部分原因是G.parasuis转化效率较低以及基因编辑工具缺乏,使得其遗传操作相对较难[6-7]。目前,应用于G.parasuis的遗传操作系统是基于同源重组原理,通过自然转化带有抗性基因的自杀质粒敲除/敲入目的基因[7-9]。该方法产生的突变株带有抗生素抗性基因,这对缺失株本身会带来极性效应,而且由于抗性基因的引入使其不可以作为弱毒疫苗使用。虽然有报道在G.parasuis基因组敲入负选择标记sacB可实现抗性基因的消除,但该方法需要构建两个质粒和进行两次自然转化[10],而且sacB基因本身易于自发失活[11-12],由此而产生的假阳性转化子也给突变株的筛选带来困难。因此对于G.parasuis,急需建立一个便捷高效的无抗性标记的基因缺失系统,以对其致病机制、毒力因子等方面进行更深入的研究。
Flp-FRT系统最初发现酿酒酵母(Saccharomycescerevisiae)的2-μm质粒,由Flp重组酶(flippase)和FRT位点组成[13]。Flp位点特异性重组酶可以识别一段48 bp的FRT(flippase recognition target) 位点序列,在两个方向相同的FRT位点间发生切割和再连接,导致位点中间片段缺失,并在作用位点留下一个FRT位点[14]。鉴于Flp-FRT系统这一特性且其具有低细胞毒性、高重组效率等优点,目前该系统被广泛应用于从原核生物[如大肠杆菌(Escherichiacoli)[15]、蓝藻(cyanobacteria)[16]等]到真核生物[如玉米[17]、黑腹果蝇[18]、哺乳动物[19]等]各种不同物种的遗传操作中。
虽然Flp-FRT系统已在多种细菌中得到广泛应用[15, 20-22],但在G.parasuis中尚未见报道。要将Flp-FRT系统应用于G.parasuis中实现多基因敲除,既要实现Flp重组酶的调控表达,也要使Flp表达质粒易于消除。目前已有各种不同的表达调控系统得到广泛研究,如来源于E.coli的阿拉伯糖操纵子[23]和乳糖操纵子[24]、来源于λ噬菌体的λcI857/PRM/PR表达调控系统[25]等,其中λcI857 /PRM/PR表达调控系统对下游基因的表达由温度调控,相较于其他表达调控系统,该系统具有经济安全等优点,下游基因表达在温度低于30 ℃时被阻遏,而温度高于42 ℃时则被激活[13]。该方法的问题在于大多数诱导型启动子无法做到完全的严谨调控,且G.parasuis的最适生长温度在37 ℃左右,若调控不够严谨,可能导致打靶质粒在整合至基因组前,就在Flp重组酶作用下发生分子内重组从而影响其转化效率。但是已有报道突变的λcI857 /PRM/PR表达调控系统在37 ℃左右也可实现严谨调控[26],并且突变的FRT位点可在一定程度上降低Flp重组酶介导的位点特应性重组效率[27-28],这或许可对解决这一问题提供帮助。
基于此,本研究首次将Flp-FRT系统应用于G.parasuis的遗传操作,将Flp重组酶基因置于该诱导型启动子调控下表达,构建了由λcI857/PRM/PR表达调控系统控制的Flp重组酶表达载体质粒,通过电转化导入G.parasuis细胞,随后在基因组上引入FRT位点以实现连续的基因敲除,利用其构建了G.parasuis的nanH、apd双基因缺失株。进一步对这一系统进行优化,通过将筛选到的突变的FRT位点与野生型FRT位点结合使用,在一定程度上降低重组效率以提高转化效率,从而提高系统稳定性。
1 材料与方法
1.1 主要试剂
胰蛋白胨大豆琼脂(trypton soy agar, TSA),胰蛋白胨大豆肉汤(trypton soy broth, TSB)购自Difco。新生牛血清(NCS)购自浙江天杭生物科技股份有限公司;烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide, NAD)、卡那霉素、庆大霉素购自德国BioFroxx公司;环磷酸腺苷(cyclic adenosine monophosphate, cAMP)购自北京索莱宝科技有限公司;蛋白胨、酵母粉、琼脂粉购自英国OXOID公司。Phanta®Max Super-Fidelity DNA Polymerase、2 × Rapid Taq Master Mix、ClonExpress II One Step Cloning Kit、ClonExpress MultiS One Step Cloning Kit购自南京诺唯赞生物科技股份有限公司;质粒DNA提取试剂盒、DNA凝胶回收试剂盒购自美国Omega Bio-Tek公司;SDS-PAGE凝胶快速制备试剂盒 (12.5%)、SDS-PAGE蛋白上样缓冲液 (5×) 购自上海碧云天生物技术有限公司;PVDF膜 (0.45 μm) 购自德国Merck公司;GAPDH Mouse McAb购自美国Proteintech Group公司;HRP Goat Anti-Mouse IgG (H+L)购自武汉爱博泰克生物科技有限公司;底物显色试剂盒(Clarity Western ECL Substrate)购自美国Bio-Rad公司;其他试剂均为国产或进口分析纯。抗Apd和抗NanH小鼠血清由本实验室制备并保存。
1.2 菌株培养与缓冲液
本研究所用菌株如表1所示,其中G.parasuisCF7066及突变株生长用TSA或TSB培养基。按照说明书将培养基灭菌后每1 L加入新生牛血清50 mL、 1%的烟酰胺腺嘌呤二核苷酸2 mL,按需加入终浓度为50 μg·mL-1的卡那霉素或20 μg·mL-1的庆大霉素。电转化所用Sucrose/glycerol (SG) buffer包含15%甘油、272 mmol·L-1蔗糖。大肠杆菌DH5α的培养使用LB肉汤或LB琼脂,根据转化质粒的抗性,加入终浓度为50 μg·mL-1的卡那霉素或20 μg·mL-1的庆大霉素。
1.3 主要质粒构建
研究中使用的质粒如表1所示,其中质粒pCP20和pKD4来自于大肠杆菌λ缺失突变系统研究[15],质粒pSHG5、pKF-ΔnanH、pKF-Δapd2和pXKN-Flp均由本课题组前期构建,质粒pSHG5C-Flp、pXKC-Flp、pKFM-Δapd的构建方法如下。
pSHG5C-Flp的结构如图1a所示,DNA片段pUCori-GmR-pA13ori使用引物CFLP-V-F/R(表2)以质粒pSHG5为模板通过PCR扩增获得,DNA片段λrepressor(cI857)-λPR-flp使用引物CFLP-F/R以质粒pCP20[29]为模板扩增,以上两个DNA片段回收后通过ClonExpress II One Step Cloning Kit试剂盒融合构建质粒pSHG5C-Flp。该质粒用于表达Flp重组酶从而介导抗性突变株中KanR表达盒的消除,其含有pA13ori温敏的复制原点,在培养G.parasuis时,30 ℃可以保持,42 ℃会发生丢失。

表1 本研究所用菌株和质粒
pXKC-Flp结构如图1b所示,DNA片段λrepressor(cI857)-λPR-flp使用引物KCFLP-V-F/R(表2)以质粒pSHG5C-Flp为模板通过PCR扩增获得, DNA片段FRT-pUCori-FRT-KanR使用引物KCFLP-F/R(表2)以质粒pXKN-Flp为模板扩增,以上两个DNA片段胶回收后通过ClonExpress II One Step Cloning Kit试剂盒融合构建质粒pXKC-Flp。该质粒用于筛选突变的mFRT位点。
pKFM-Δapd结构如图1c所示,apd上下游同源臂以CF7066基因组DNA为模板扩增,pUC ori和卡那霉素抗性表达盒的DNA片段使用引物KFM-V-F/R(表2)以质粒pKF-Δapd2为模板扩增,DNA片段mFRT使用引物KFM-F/KFM-R(表2)以质粒pXKC-Flp为模板扩增,以上4个DNA片段回收后通过ClonExpress II One Step Cloning Kit试剂盒融合构建质粒pKFM-Δapd。
1.4 自然转化和电转化
G.parasuis自然转化参考 Bigas等[8]的方法。首先将细菌接种于TSA平板均匀划线并于37 ℃培养16 h,长出的菌苔用2 mL TSB洗下并调整OD600 nm约0.9,取20 μL菌液、20 μL cAMP (8 mmol·L-1)、1 μg质粒混匀后滴于TSA平板正置于37 ℃培养5 h。取200 μL TSB洗下菌苔并均匀涂布于相应抗性的TSA平板,于37 ℃培养。
G.parasuis电转化参考Lancashire等[30]的方法并稍作改进。细菌接种于TSA平板均匀划线并于37 ℃培养14 h,长出的菌体用2 mL SG buffer洗下。离心收集菌体后用2 mL SG buffer重悬洗涤,重复3次,最终调整OD600nm约2.0。取100 μL菌液加入5 μg质粒,冰浴30 min后加入预冷的0.2 cm电转杯中,于2.5 kV电击一次后加入1 mL TSB,在37 ℃转速为200 r·min-1的摇床复苏2 h后均匀涂布于相应抗性的TSA平板。由于穿梭质粒pSHG5C-Flp携带温度敏感型复制子,其在30 ℃可复制,而温度高于42 ℃则停止复制[31]。电转后细菌的培养条件为37 ℃/30 ℃交替培养,温度每12 h变换1次,以维持转化子中温敏质粒复制。
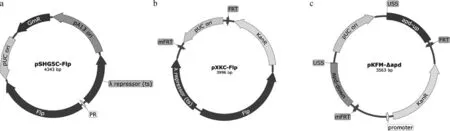
a. 温敏穿梭质粒pSHG5C-Flp,表达Flp重组酶介导Kan抗性基因消除;b. 质粒pXKC-Flp,通过转化大肠杆菌DH5α后的自剪切筛选mFRT位点;c. 用于自然转化的自杀质粒pKFM-Δapd,单侧含有mFRT位点a. The temperature-sensitive shuttle plasmid pSHG5C-Flp that mediated antibiotic resistance gene excision by Flp recombinase; b. The plasmid pXKC-Flp including the mutated FRT site, which was screened according to the self-excised occurrence in the transformed DH5α cells; c. The suicide plasmid pKFM-Δapd containing mFRT in unilateral site that was used for natural transformation图1 本研究所用质粒图谱Fig.1 The maps of plasmids used in the study

表2 本研究所用引物
1.5 Flp重组酶作用检测
用自杀质粒pKF-Δapd2自然转化G.parasuisCF7066,转化菌体涂布到TSA/KanR平板上于37 ℃培养,长出的单菌落用引物APD-OUT-F/R(表2)进行PCR筛选,得到CF7066Δapd::KanR突变体。接下来,用Flp表达质粒pSHG5C-Flp电转化CF7066Δapd::KanR,转化菌体涂布到TSA/GmR平板上在37 ℃/30 ℃交替培养,再使用上述引物进行PCR扩增,筛选并鉴定无抗缺失株CF7066Δapd,通过抗性基因消除确定Flp酶的作用。
1.6 nanH和apd基因连续敲除
双基因缺失株构建流程如图2a所示,构建过程中CF7066基因组基因型变化如图2b所示。首先用Flp表达质粒pSHG5C-Flp电转化G.parasuisCF7066,涂布于TSA/GmR平板上于37 ℃培养,长出的转化子使用质粒鉴定引物G5C-F/R(表2)进行PCR扩增,检测抗性平板上长出的转化子是否含有质粒pSHG5C-Flp,含有质粒的重组菌株即为CF7066(pSHG5C-Flp)。然后用pKF-ΔnanH自然转化CF7066(pSHG5C-Flp),涂布TSA/KanR平板上于37 ℃/30 ℃交替培养,长出的转化子进行菌落PCR,使用基因nanH外侧引物NANH-OUT-F/R和质粒鉴定引物G5C-F/R,筛选出无抗缺失株CF7066ΔnanH(pSHG5C-Flp)。
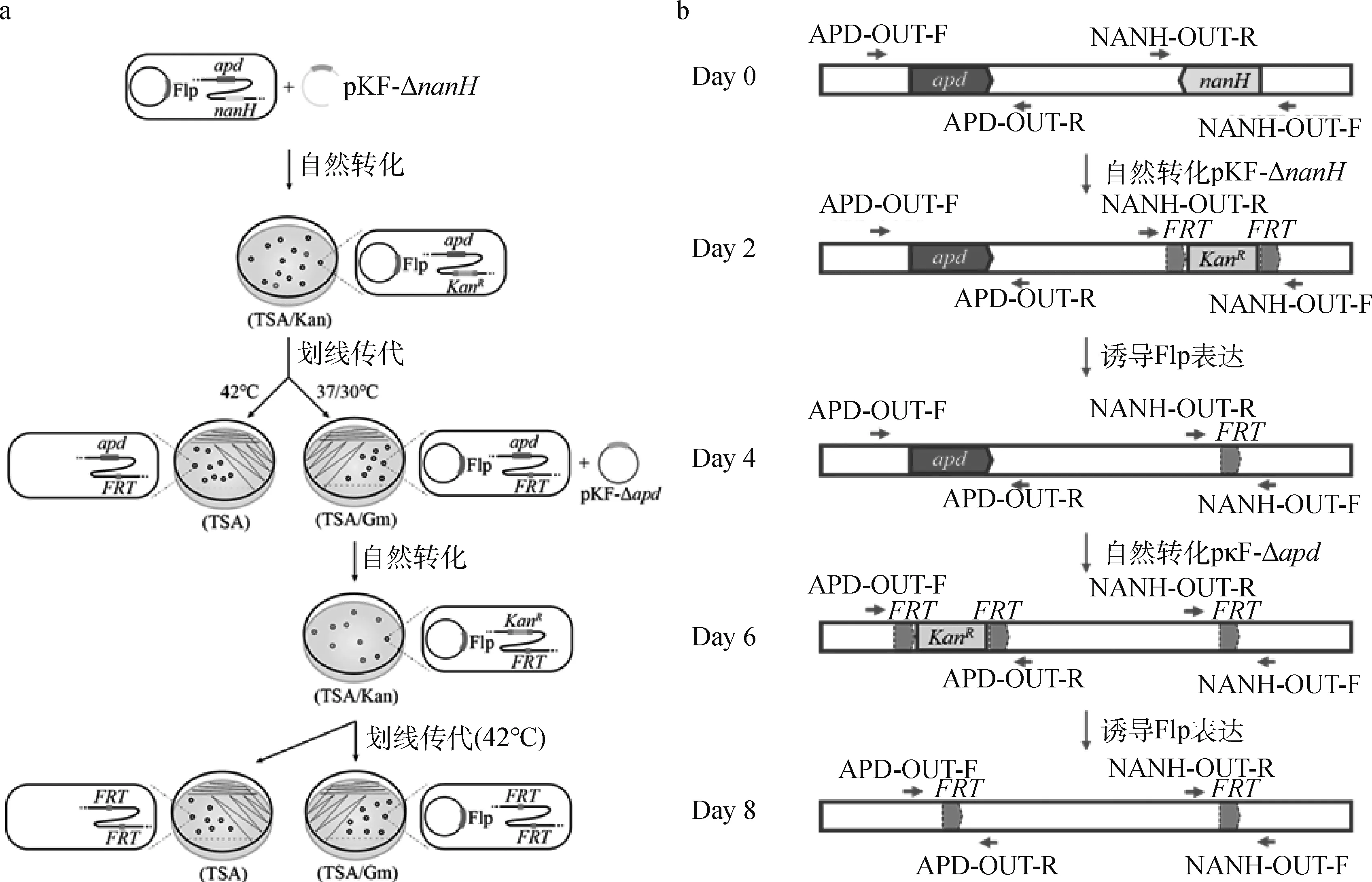
a. 双基因敲除流程图;b.双基因敲除基因型变化示意图a. Schematic diagram of double gene knockout; b. Genotype changes of CF7066 genome图2 通过质粒pSHG5C-Flp进行双基因敲除的策略Fig.2 The strategy of double gene knockout by using plasmid pSHG5C-Flp
接下来,将筛选到的有抗缺失突变株于TSA平板划线传代,在42 ℃下培养以消除质粒pSHG5C-Flp,获得无抗单基因缺失株CF7066ΔnanH;同时将其于庆大霉素抗性平板传代,在37 ℃/30 ℃交替培养,温度每12 h变换1次,以保证温敏穿梭质粒pSHG5C-Flp的复制和Flp酶表达,得到抗性消除的单基因缺失株CF7066ΔnanH(pSHG5C-Flp)。再用自杀质粒pKF-Δapd2自然转化CF7066ΔnanH(pSHG5C-Flp),用apd外侧引物APD-OUT-F/R和质粒鉴定引物G5C-F/R(表2)通过筛选无抗双基因缺失株CF7066ΔnanHΔapd(pSHG5C-Flp),最后在TSA平板上42 ℃培养以消除质粒,获得双基因缺失株CF7066ΔnanHΔapd。
祖母信奉的瓜达卢佩圣母,代表着与西班牙殖民文化相对的墨西哥本土文化,是墨西哥的文化身份的象征。(石平萍,2005:25)赛利亚对祖母态度的变化在本质上也是对祖母所代表的墨西哥传统文化的态度变化。祖母长期被赛利亚视为“可怕祖母”。在叙述祖母的故事中,她逐渐理解了祖母严苛的外表下对儿孙们的爱,感觉到了与祖母之间的联系:“我就是可怕祖母,我成为她,看到了她的内心。”(424)
1.7 Western blot
使用重组Apd和抗NanH蛋白的小鼠抗血清,通过Western blot检测NanH和Apd蛋白的表达。野生株和缺失株分别接种于TSA平板37 ℃过夜培养,取等量菌体用40 μL双蒸水重悬,加入10 μL SDS-PAGE蛋白上样缓冲液 (5×) 并煮沸10 min后置于冰上冷却。样本进行SDS-PAGE凝胶电泳并转移到PVDF膜上,TBST洗涤3次,于含5%脱脂乳的TBST溶液室温封闭6 h后,分别按1∶5 000 加入抗Apd、抗NanH血清和GAPDH单克隆抗体于4 ℃过夜孵育。TBST洗涤3次后,将PVDF膜放入含5%脱脂乳的TBST溶液中,分别按1∶5 000加入HRP标记的羊抗鼠IgG,室温孵育1 h,TBST洗涤3次,于ECL超敏化学发光液中显色后将PVDF膜置于凝胶成像系统中显色。
1.8 转化效率比较试验
转化效率比较试验按照“1.6”中缺失第一个基因的方法进行。首先电转化获得组菌株CF7066(pSHG5C-Flp)自然转化方法进行,然后取1 μg质粒pKF-Δapd2和pKFM-Δapd,分别自然转化CF7066(pSHG5C-Flp)。每组设置3个重复,待长出转化子后分别统计转化子数。
1.9 生长曲线测定
使用 Bioscreen 全自动生长曲线分析仪测定菌株生长曲线,野生株和缺失株分别于TSA平板活化三代后取单菌落接种于5 mL TSB培养基,于37 ℃ 200 r·min-1震荡培养12 h后按1∶100转接于新鲜的TSB培养基。取Bioscreen全自动生长曲线分析仪专用的检测板,每孔加入200 μL转接菌液,每30 min测定菌液在600 nm波长处的吸光度(OD600nm),记录并绘制生长曲线。
2 结 果
2.1 Flp重组酶消除基因组上KanR基因
为了证实Flp-FRT系统在G.parasuis中是否发挥作用,本研究用λ噬菌体的可调控启动子cI857/PRM/PR调控Flp基因的表达,构建了质粒pSHG5C-Flp(图1a)。然后,用Flp重组酶的表达质粒pSHG5C-Flp电转化CF7066Δapd::KanR,对长出的转化子用基因apd外侧引物和质粒鉴定引物G5C-F/R通过PCR检测。在基因apd外侧引物APD-OUT-F/R的扩增13个菌落中,8个菌落的基因型为Δapd::KanR(3 041 bp),5个菌落的基因型为Δapd(1 535 bp)。而同时pSHG5C-Flp质粒鉴定引物G5C-F/ G5C-R的扩增结果显示,在8个基因型为Δapd::KanR的菌落中不存在质粒,而在5个基因型为Δapd的菌落中有质粒存在(869 bp)(图3)。该结果表明,在质粒pSHG5C-Flp得以成功转化的有抗性标记的突变株,Flp重组酶表达并介导了KanR基因的消除,从而获得了G.parasuis无抗缺失株。

a. 用Flp酶表达质粒pSHG5C-Flp电转化CF7066后基因型的改变;b.质粒pSHG5C-Flp在转化子中的存在a. The genotype change of CF7066 that was ecletrotransformed with pSHG5C-Flp expressing Flp recombinase amplication; b. The existence of pSHG5C-Flp in transformed bacterial cells图3 Flp重组酶介导的KanR基因消除Fig.3 The KanR gene excision mediated by Flp recombinase
2.2 Flp-FRT系统介导nanH和apd基因连续敲除
为了能够进行G.parasuis双基因连续敲除,首先用表达Flp酶的温敏质粒pSHG5C-Flp电转化CF7066获得重组菌CF7066(pSHG5C-Flp)。然后用自杀质粒pKF-ΔnanH自然转化重组菌CF7066(pSHG5C-Flp),获得的转化子用PCR筛选。结果表明,所检测的22个转化子中,都出现了有抗性标记的缺失基因型ΔnanH::KanR(3 923 bp),其中18个中还包含抗性消除的缺失基因型ΔnanH(2 417 bp), 而且在出现无抗缺失株的18个转化子中均检测到了Flp酶表达质粒的存在(图4a)。选择1#转化子在TSA平板分别于42 ℃和37 ℃/30 ℃上培养。结果表明,在42 ℃培养的22个单菌落中,19个菌落出现了抗性基因消除,但质粒pSHG5C-Flp的丢失不一致;而在37 ℃/30 ℃培养的22个单菌落中,22个菌落中的抗性基因全部消除,并且质粒pSHG5C-Flp全部存在,选择17#菌株进行下一个基因敲除(图4b、c)。可见,CF7066(pSHG5C-Flp)可表达Flp酶并介导抗性突变株中KanR的消除,同时也证实温敏质粒pSHG5C-Flp在37 ℃/30 ℃ 培养可以表达具有活性的Flp重组酶。

a.用pKF-ΔnanH自然转化CF7066(pSHG5C-Flp)后基因型的改变;b. 42℃培养后抗性消除和质粒丢失;c. 37/30 ℃培养后抗性消除和质粒丢失a. The genotype change of CF7066(pSHG5C-Flp)transformants following natural transformation with pKF-ΔnanH; b. The resistance cassette excision and plasmid curing following culturation at 42 ℃; c. The resistance cassette excision and plasmid caring following cultaration at 37/30 ℃图4 缺失株CF7066ΔnanH构建Fig.4 The construction of the deletion strain CF7066ΔnanH
接下来,再用质粒pKF-Δapd2自然转化CF7066ΔnanH(pSHG5C-Flp)。结果显示,虽然在22个菌落中均可检测到质粒pSHG5C-Flp,但仅有3个菌落出现了有抗性标记的apd缺失的基因型,而其余19个为转化前的基因型(图5a)。将筛选到的有抗性消除的apd基因缺失株(11#)于42 ℃ 在TSA平板上培养,筛选并获得到了双基因缺失株CF7066ΔnanHΔapd(12#),同时也实现了质粒pSHG5C-Flp的消除(图5b)。以该双基因缺失株为模板,用nanH和apd基因外侧引物扩增目的片段并进行序列分析,结果表明示nanH和apd均成功敲除,并在缺失的基因座留下了FRT位点(图5c)。进一步,分别使用Apd和抗NanH蛋白的小鼠抗血清进行 Western blot检测,可见双基因缺失株中无两种蛋白的印迹,而野生型菌株中均有两种蛋白预期大小的印迹(图5d)。上述结果表明,在CF7066菌株中预先转化表达Flp重组酶的温敏质粒pSHG5C-Flp,可以实现G.parasuis基因组上KanR基因的切除以及双基因连续敲除。
2.3 FRT突变位点的筛选
在进行nanH和apd基因连续敲除时,有抗性标记的突变体在第一个基因敲除时出现的比率为100%(22/22)(图4a),而在第二个基因敲除时出现的比率仅为13.6%(3/22)(图5a)。抗性突变体在敲除第二个基因时的比率大为下降,有可能是质粒pSHG5C-Flp在菌体内的持续存在和Flp重组酶持续表达的积累,对自然转化后的自杀质粒介导位点特异性重组,从而减少了抗性缺失突变株的获得。因此,通过筛选有点突变FRT的序列,以降低积累的Flp重组酶对自然转化后质粒介导的同源重组的干扰和抗性突变株的获得。
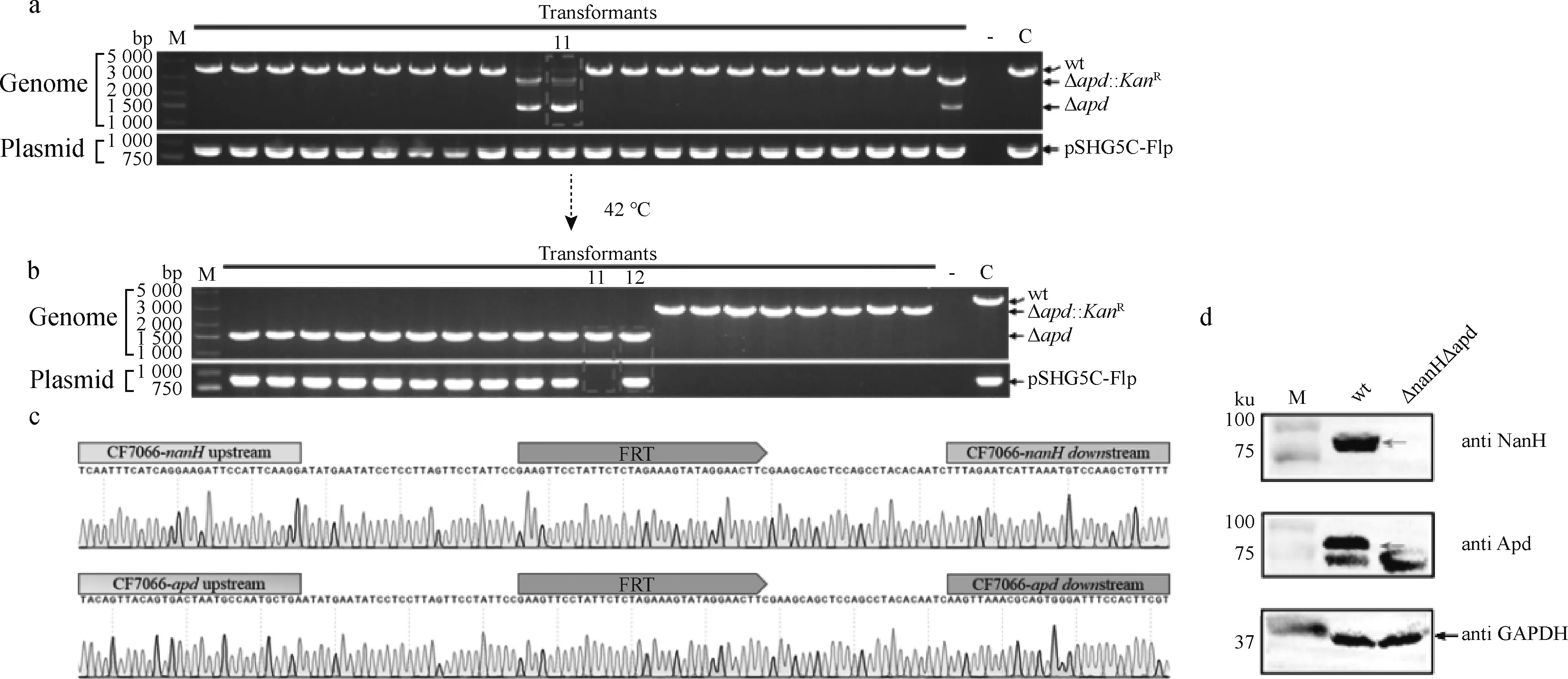
a. 用pKF-Δapd2自然转化CF7066ΔnanH(pSHG5C-Flp)后基因型的改变;b. 42 ℃培养后抗性消除和质粒丢失;c. PCR产物测序鉴定靶基因的敲除;d. Western blot 检测敲除基因蛋白的表达a. The genotype change of CF7066ΔnanH(pSHG5C-Flp)transformants following natural transformation with pKF-Δapd2; b. The resistance cassette excision and plasmid curing following culturation at 42℃; c. Identification of gene knockout by sequence analysis of PCR products; d. Detection of the protein expression of the knocked-out genes图5 缺失株CF7066ΔnanHΔapd的构建Fig.5 The construction of the deletion strain CF7066ΔnanHΔapd

a. mFRT筛选结果;b. mFRT测序结果(wtFRT.野生型FRT位点;mFRT.突变FRT位点)a. The screening results of mFRT; b. The mFRT sequence (wtFRT. Wild type FRT; mFRT. Mutant FRT)图6 突变FRT位点的筛选Fig.6 Screening of the mutated FRT site
2.4 突变FRT位点提高系统的稳定性
首先,将质粒pKF-Δapd2的KanR基因上游FRT位点替换为筛选到的突变FRT位点(mFRT),构建质粒pKFM-Δapd(图1c)。然后,用pKFM-Δapd自然转化单基因缺失株CF7066ΔnanH(pSHG5C-Flp),结果在22个菌落中,均可检测到质粒pSHG5C-Flp的存在并出现了有抗性标记的apd基因缺失的基因型(Δapd::KanR)(图7a)。可见,单侧突变的mFRT位点有效降低了Flp重组酶的负效应,使有抗性标记突变体出现的比率从原来的13.6%(3/22)(图5a)提高到100%(22/22)。最后,选择7#突变体于TSA/Gm平板划线传代,并于37 ℃/30 ℃交替培养,22#菌落的抗性表达盒被切除(图7b)。以上结果表明,抗性基因一侧引入突变的mFRT位点,保证了自然转化后同源重组和等位交换的发生,而提高了该系统的有效性和稳定性。此外,用质粒pKF-Δapd2和pKFM-Δapd分别转化CF7066(pSHG5C-Flp)进行了转化效率的比较。结果表明,使用具有mFRT位点的质粒pFKM-Δapd转化时,获得的转化子数目比未突变时提高约6倍(图7c)。

a. 用pKFM-Δapd自然转化CF7066ΔnanH(pSHG5C-Flp)后基因型的改变;b. 37/30 ℃培养后抗性消除和质粒丢失;c. 质粒pKF-Δapd2和pKFM-Δapd转化效率比较,***代表P<0.001a. The genotype change of CF7066ΔnanH(pSHG5C-Flp) transformants following natural transformation with pKFM-Δapd; b. The resistance cassette excision and plasmid curing following culturation at 37/30 ℃; c. Comparison of the transformation efficiency of plasmid pKF-Δapd2 and pKFM-Δapd,*** represents P<0.001图7 突变FRT位点的应用Fig.7 The employment of mutant FRT site
2.5 nanH、apd基因缺失对CF7066生长的影响
比较野生株CF7066和3株缺失株ΔnanH、Δapd、ΔnanHΔapd的生长曲线发现,Δapd缺失株与野生株的生长曲线并无显著差异;而CF7066缺失株ΔnanH和ΔnanHΔapd与其野生型相比,不仅进入对数期的时间更晚、稳定期的时间更短,到达稳定期的OD600nm数值也都低于野生株(图8)。以上结果说明apd基因的缺失对G.parasuis生长速率并无显著影响,而nanH基因的缺失显著降低了生长效率,并使得菌株更早进入衰亡期。因此CF7066唾液酸酶nanH基因的缺失可能导致环境中唾液酸代谢障碍从而影响CF7066的生长。
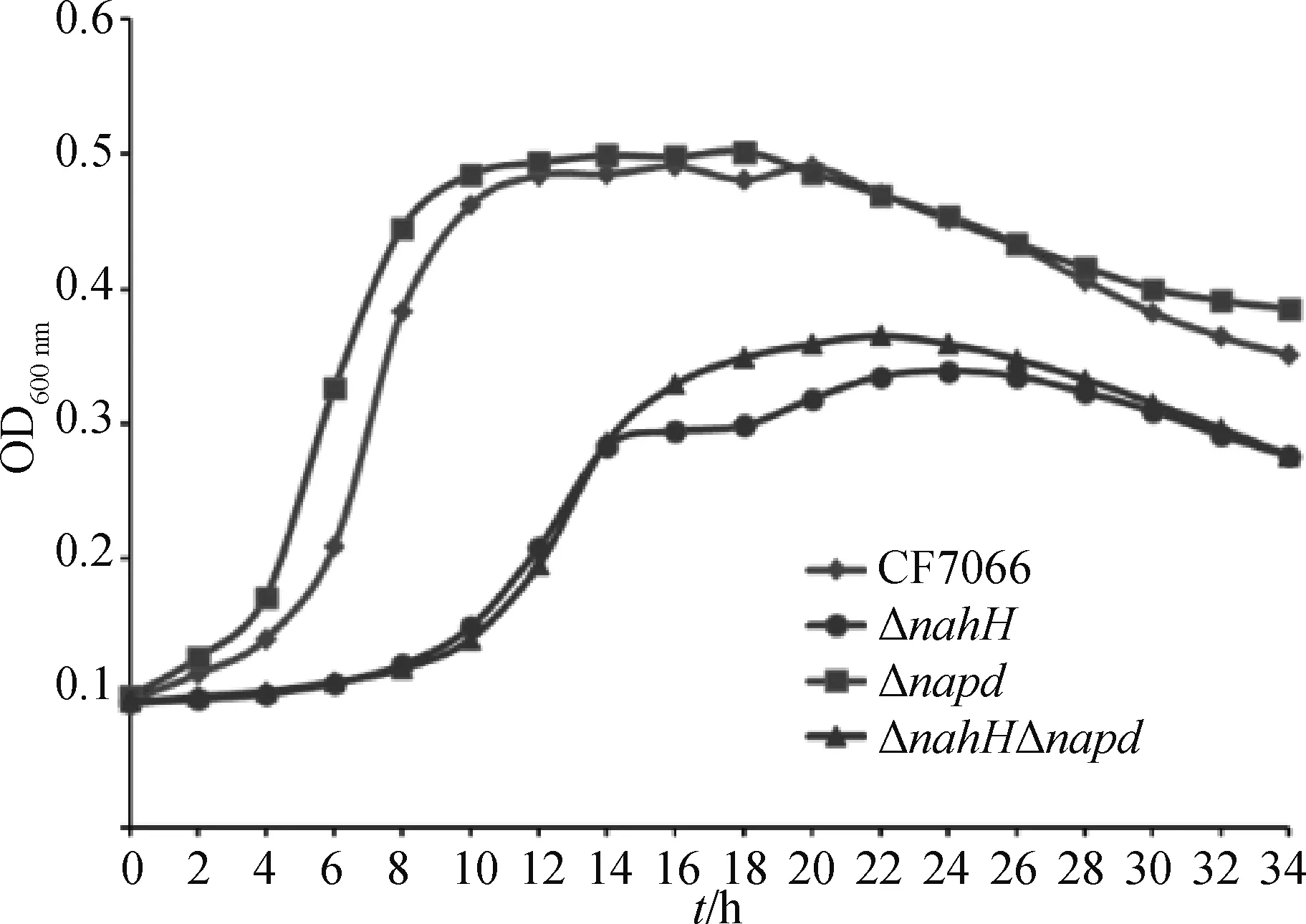
图8 CF7066野生型和基因缺失株的生长曲线Fig.8 The growth curves of the CF7066 WT and deletion mutants
3 讨 论
G.parasuis遗传操作工具的缺乏限制了其分子生物学研究的进展,因此建立一个稳定高效的遗传操作系统对其致病机制、毒力因子等研究具有重要意义。本研究利用λcI857/PRM/PR表达调控系统控制Flp重组酶表达,随后自然转化自杀性质粒在基因组靶位点引入含有FRT位点的抗性标记,通过Flp重组酶表达切除抗性基因,实现了抗性标记循环使用和基因连续敲除,最后提高温度消除Flp表达质粒,成功敲除了G.parasuisCF7066的nanH和apd基因。
对Flp重组酶的表达质粒pSHG5C-Flp而言,Flp的表达受到λ噬菌体右启动子PR控制和cI857/PRM蛋白的调控,即该启动子在30 ℃被阻遏,在42 ℃ 被激活[13]。作者计划用Flp酶表达质粒电转化G.parasuis后在30 ℃培养,然后用自杀性质粒自然转化后也在30 ℃培养并获得抗性突变体,因为在30 ℃培养,温敏质粒可以稳定复制,而且Flp酶没有活性。最后,对自然转化获得的抗性突变株,在42 ℃培养以诱导Flp酶的表达进而切除抗性基因并且消除质粒。然而实际情况是,由于G.parasuis在30 ℃生长缓慢,在此温度下多次自然转化均无转化子长出。已有研究报道突变的λcI857/PRM/PR系统可将调控表达的温度从30 ℃提高至37 ℃[26]。所以在当前研究中,为了既调控Flp重组酶的表达又不影响自然转化效率,作者将携带CF7066(pSHG5C-Flp)在37 ℃/30 ℃交替培养作为受体菌。结果,在自然转化敲除CF7066的nanH和apd基因时,初代转化子中竟然出现了无抗性标记缺失突变体,一次性完成了同源重组和抗性消除(图4、5)。尽管自然转化初代可获得无抗缺失突变体令人欣喜,但在敲除第二个基因时,自然转化后发生同源重组的突变体比率明显下降。所幸的是,通过构建单侧FRT位点突变的质粒,有效提高了自然转化后同源重组的发生和抗性突变体的产生,从而保证了该双基因敲除系统的有效性和稳定性。
G.parasuis中遗传操作方法比较有限,本研究与Bigas等[8]报道的遗传操作系统相比,本方法消除了遗留在基因组上的抗性基因表达盒,减少了潜在引起的极性效应,更有利于对目的基因的进一步研究。另外,Zhang等[10]也报道了利用反选择标记进一步敲除抗性基因的方法,但该方法每敲除一个基因需要构建两个质粒,而且由于大部分G.parasuis对蔗糖不敏感、sacB基因本身易于自发失活而产生假阳性转化子,这使得该方法难以在更多的菌株中使用。而本研究所建立的方法,不论在操作的简便性和对转化菌株的普适性上都有所提高,而且还可以进行双基因连续敲除。
对G.parasuis而言,广泛存在的限制修饰系统引起的转化屏障是其遗传操作困难的原因之一[6-7]。本研究只需要将表达Flp重组酶的温敏穿梭质粒进行一次电转化,就可以实现两个基因的连续敲除,这不仅降低了操作难度也极大简化了实验流程。进一步对该系统进行优化,通过筛选突变的FRT位点降低Flp重组酶介导的重组效率,虽然在一定程度上降低了自然转化后初代转化子中出现无抗敲除的效率,但极大提高了系统的稳定性。最终获得了可以用于G.parasuis的无抗和双基因连续敲除的突变系统。
所不足的是,本研究利用Flp-FRT系统实现抗性基因的敲除,最终会在目的基因处留下一个FRT位点,因此尚难以实现不留痕迹的基因敲除。另一方面,留下的FRT位点也给连续的多基因敲除带来一定困扰,虽然本研究成功利用Flp-FRT系统构建了CF7066的双基因敲除菌株,这可能是由于基因nanH和apd在基因组上相距较远(约913 kb),如果发生基因nanH和apd间大DNA片段敲除必然会导致中间必须基因的失活造成致死性突变。但在敲除两个相隔较近的基因时,Flp-FRT系统的这一缺陷是不得不考虑的问题。为了避免这一问题的发生,可以在敲除距离较近的两个基因时分别使用不同的两组FRT位点,从而消除该系统的这一缺陷[27-28,32]。
4 结 论
利用Flp-FRT位点特异性重组系统,构建了副猪嗜血杆菌CF7066双基因缺失株,建立了针对副猪嗜血杆菌的无抗性标记的基因缺失系统,为副猪嗜血杆菌基因功能研究、基因工程疫苗研发提供了有效的工具。
