变应性鼻炎特异性病毒应答相关基因的生物信息学分析
2022-03-07曹玉洁李正埼周纯李华斌
曹玉洁,李正埼,周纯,李华斌
(1.复旦大学附属眼耳鼻喉科医院 耳鼻咽喉科,上海 200031; 2.中山大学附属第一医院耳鼻咽喉科医院 广州市耳鼻咽喉科学重点实验室,广东 广州 510080)
变应性鼻炎(allergic rhinitis,AR)是一种IgE介导的鼻黏膜炎症,其症状包括喷嚏、鼻痒、鼻塞和清涕,影响全球20%~40%的人群[1]。AR严重影响患者的生活质量、工作效率及学习能力,并给社会带来巨大的经济负担[2-4]。病毒感染是呼吸道感染最常见的原因之一,其中以鼻病毒最为典型[5]。气道上皮细胞作为病毒感染和复制的宿主细胞利用模式识别受体识别病毒的病原体相关分子模式,并启动下游免疫应答。模式识别受体包括toll样受体(toll-like receptors, TLR)3,它可以识别病毒双链RNA(dsRNA)及其合成类似物poly (I:C),以诱导细胞因子、趋化因子和I型干扰素的表达[1,6-7]。鼻病毒的dsRNA在复制过程中产生,然后被TLR3识别[8]。研究报道,病毒感染可导致AR的发生和加重[9]。Greiff等[10]发现鼻病毒感染可激活嗜酸性粒细胞和中性粒细胞以加重呼吸道炎症。在社区获得性感冒的急性期,AR患者的嗜酸性粒细胞水平升高[11]。Skoner等[12]也发现在鼻病毒-39刺激后,AR患者的白细胞、辅助T淋巴细胞、抑制型T淋巴细胞以及自然杀伤细胞的数量和功能与非AR患者相比均发生变化,这可能来源于先前存在的气道过敏炎症。此外,在病毒性感冒期间,AR患者的CT评分显著高于非AR患者,鼻气流和黏液纤毛清除率则较低[13-14]。综上所述,鼻病毒感染的AR患者病情与气道炎症更为严重。然而,鼻病毒加重气道炎症的机制仍不清楚。为进一步了解鼻病毒感染导致AR加重的分子机制,本研究通过生物信息学方法筛选出dsRNA刺激后AR鼻黏膜上皮细胞特异性差异表达基因 (differentially expressed genes,DEGs)。然后通过GO和KEGG进行通路富集分析,以确定这些基因参与的生物学过程及功能。此外,通过STRING数据库构建蛋白质相互作用(protein-protein interaction,PPI)网络,寻找AR特异性的关键基因和集簇。
1 材料与方法
1.1 数据来源
从GEO数据库(http://www.ncbi.nlm.nih.gov/geo/)中下载基于GPL13158平台的GSE51392数据集。共选取11例受试者进行进一步分析,包括5例AR患者和6例健康对照受试者(healthy control,HC)。分离培养受试者鼻黏膜上皮细胞,poly (I:C)刺激24 h后提取RNA并使用微阵列(affymetrix U133+ PM genechip array)进行分析[15]。
1.2 原始数据预处理及DEGs筛选
使用R语言的affy包,采用robust multi-array average(RMA)算法对原始数据进行背景校正和标准化[16-17]。根据注释信息将探针ID映射到基因符号,如果多个探针对应于同一基因符号,则对这些探针的表达值取平均值。本研究将数据分为3组进行比较,分别是AR患者来源上皮细胞受dsRNA刺激前后的差异比较,HC来源上皮细胞受dsRNA刺激前后的差异比较,以及未刺激状态下HC与AR患者来源上皮细胞之间的差异比较。使用limma包和经验贝叶斯方法筛选DEGs[15, 17-18]。采用false discovery rate(FDR)对P值进行校正。筛选标准为校正后P<0.05。根据校正后P值和倍数变化(fold change,FC)的对数值绘制火山图。利用UNIPROT数据库(https://www.uniprot.org/)获取基因的详细注释和功能[19]。
1.3 GO和KEGG通路富集分析
为了确定筛选出的DEGs参与的生物学过程及功能。本研究通过DAVID数据库进行GO和KEGG通路富集分析。DEGs的GO功能富集包括生物过程、细胞成分和分子功能3个层次。筛选标准为P<0.05。
1.4 PPI网络构建和重要集簇选择
使用STRING在线分析工具(search tool for retrieval of interactions genes,http://www.string-db.org/)以联合评分≥0.700为临界值构建PPI网络。将PPI网络结果重新加载到cytoscape软件(version 3.7.2)中进一步分析。在网络中,每个节点代表一个基因,连线代表节点之间的连接。Degree表示基因节点之间的连接或相互作用。Degree较高的节点被认为具有重要生物学功能的hub基因。使用cytoscape的分子复合物检测插件(molecular complex detection,MCODE)从PPI中提取重要基因集簇。筛选标准为:“degree cutoff=2”,“node score cutoff=0.2”,“k-core=2”,“max. depth=100”。此外,在cytoscape中使用Glue GO插件对各集簇的基因进行GO富集分析。
2 结果
2.1 dsRNA刺激后的AR特异性DEGs
通过筛选发现,HC来源上皮细胞经dsRNA刺激后共9 550个DEGs,其中4 214个上调DEGs,5 336个下调DEGs。与基线水平相比,AR来源上皮细胞经dsRNA刺激后共检测到8 578个DEGs,其中3 605个上调,4 973个下调。而在dsRNA刺激前,未发现HC与AR的DEGs。为了进一步探究AR上皮细胞对dsRNA的特异性应答反应,我们通过韦恩图对3组DEGs进行分析。共筛选出545个上调和400个下调的AR特异性DEGs。表1展示了根据差异倍数排列的前10个上调或下调的AR特异性DEGs。包括上调的血小板碱性蛋白(platelet basic protein,PPBP),又称C-X-C motif趋化因子7(CXCL7)以及下调的白细胞介素20(interleukin-20,IL-20)、B细胞连接蛋白(B-cell linker protein,BLNK)、CCAAT/增强剂结合蛋白delta(CCAAT/enhancer-binding protein delta,CEBPD)和淋巴细胞抗原96(lymphocyte antigen 96,LY96)。见图1、2。

图1 两组上皮细胞分别受dsRNA刺激前后的DEGs火山图 1A:HC; 1B:AR 红点和蓝点分别表示上调和下调的DEGs,黑点表示基因没有显著差异 图2 两组上皮细胞分别受dsRNA刺激前后以及基线水平HC和AR的DEGs的维恩图 2A:上调基因; 2B:下调基因

表1 dsRNA刺激后差异变化前10位的AR特异性上调或下调的基因
2.2 DEGs的GO和KEGG富集分析
为了进一步明确AR特异性DEGs参与的生物学过程及功能,我们分别对上调或下调的DEGs进行GO和KEGG通路富集分析。结果图3、4所示。dsRNA刺激后,AR特异的功能和信号通路包括增强的“cell adhesion”和“defense response”,以及受损的“positive regulation of tumor necrosis factor production”。

图3 AR特异性上调基因的GO和KEGG通路分析 A:生物学过程;B:细胞成分;C:分子功能;D:KEGG通路
2.3 PPI网络分析
接下来我们使用STRING构建了AR特异性DEGs的PPI网络。该PPI网络由791个节点和603条连线组成(图5)。在PPI网络中,我们选择排名前16位的高Degree节点作为hub基因(图6),包括PPBP/CXCL7,它与中性粒细胞趋化和激活有关。通过MCODE从PPI网络中筛选出8组集簇,同时对各集簇进行GO富集分析。本研究选择综合分数高于5分的集簇进行进一步分析。共筛选5组集簇分别代表不同的生物途径。集簇1(MCODE score=11.846)包括14个DEGs富集于nuclear-transcribed mRNA catabolic process, nonsense-mediated decay通路。集簇2(MCODE score=8.6)包括11个DEGs但未发现富集的GO通路。集簇3(MCODE score=8) 包括8个DEG富集于synaptic vesicle recycling通路。集簇4(MCODE score=6.615)包括14个DEGs富集于adenylate cyclase-inhibiting G protein-coupled receptor signaling pathway通路。集簇5(MCODE score=6)包括6个DEGs但未发现富集的GO通路。

图4 AR特异性下调基因的GO和KEGG通路分析 A:生物学过程;B:细胞成分;C:分子功能;D:KEGG通路
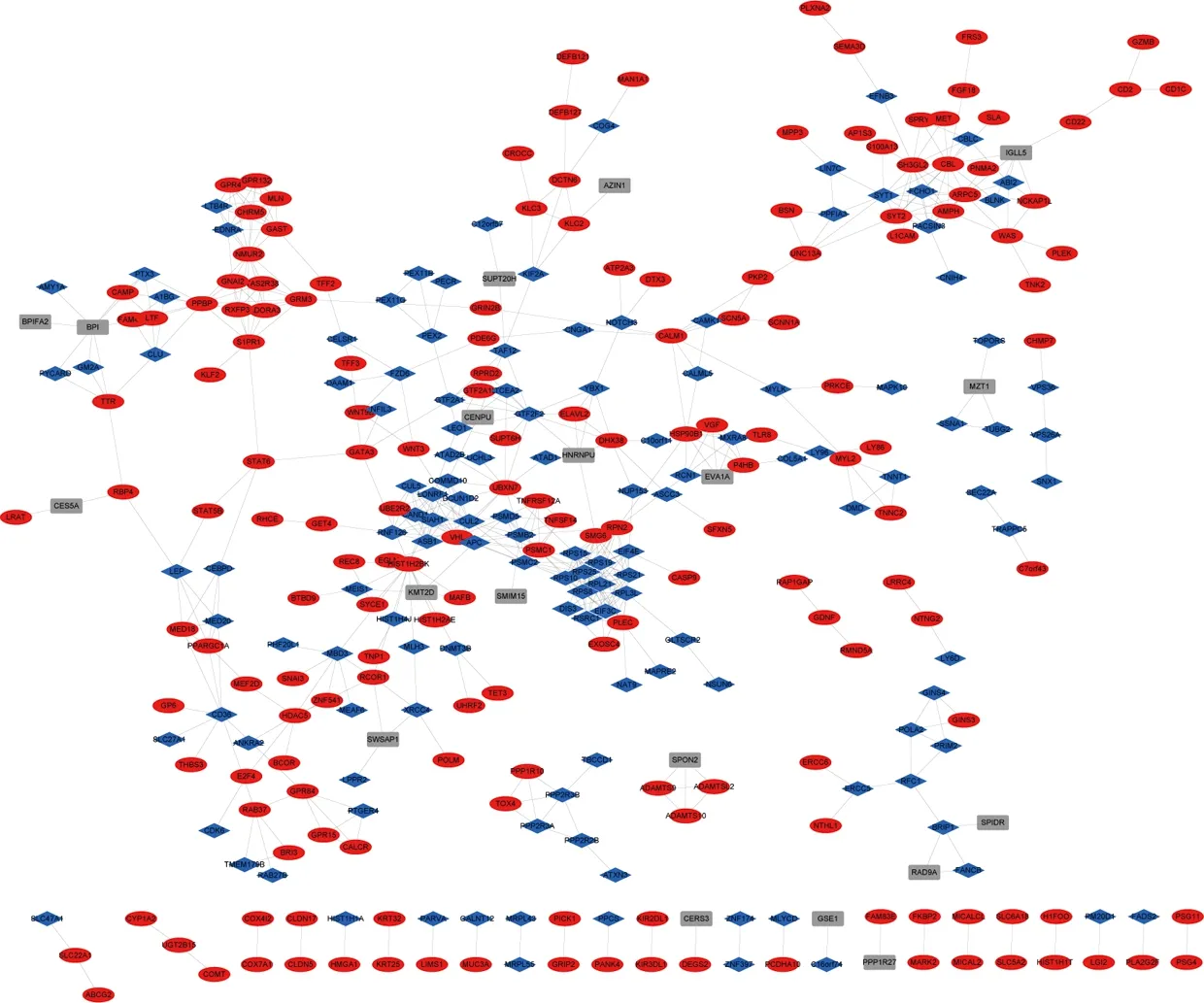
图5 蛋白质相互作用网络结果,红色代表上调DEGs,蓝色代表下调DEGs

图6 从PPI网络筛选出16个hub基因。Degree用从黄(低)到红(高)的渐变颜色表示
3 讨论
鼻黏膜上皮是呼吸道病毒感染和复制的主要部位。病毒感染导致的气道炎症加重可能是由病毒的直接损伤和宿主清除病毒这一免疫过程的间接损伤引起。病毒损伤呼吸道上皮后又可增加AR患者对变应原的敏感性,进一步加重炎症[1]。Wagener等[15]鉴定了来自同一个体的鼻黏膜和支气管上皮细胞经dsRNA刺激后的基因表达谱,并报道上、下呼吸道上皮对dsRNA感染的反应大致相同,呼吸道病毒影响线粒体基因表达。Wesolowska-Andersen等[20]通过转录组学分析发现高病毒携带者的气道免疫细胞浸润增加、纤毛基因下调和2型炎症受抑。用鼻病毒感染气-液平面分化培养的下呼吸道上皮细胞后进行RNA-seq分析,也发现一系列哮喘特异性病毒应答相关基因,包括与炎症通路、上皮结构重塑以及纤毛组装和功能相关的基因[21]。本研究比较了dsRNA刺激前后AR与HC上皮的DEGs,聚焦于dsRNA刺激后AR的特异性DEGs,包括上调基因PPBP/CXCL7以及下调基因IL-20、BLNK、CEBPD、LY96。此外,我们分析了dsRNA刺激后AR特异性DEGs相关的功能和信号通路。最后,我们从PPI网络中筛选出hub基因PPBP/CXCL7。以上结果可能有助于阐明RV感染导致AR炎症加重的分子机制。
首先,我们发现dsRNA刺激后PPBP/CXCL7在AR中特异性上调,并作为PPI网络中的hub基因。CXCL7可形成生物活性链,包括结缔组织激活肽III(connective tissue-activating peptide III,CTAP-III)和中性粒细胞激活肽2(neutrophil-activating peptide 2,nap2)。据报道,CXCL7可促进中性粒细胞粘附并穿越血管内皮,且有重要的抗菌作用[22-23]。在稳定型严重/非常严重慢性阻塞性肺病患者的支气管黏膜中CXCL7的mRNA水平、CXCL7+细胞数量和中性粒细胞数量均增加,这表明CXCL7可能是重度慢性阻塞性肺病患者支气管黏膜中性粒细胞增多的原因[24]。目前的研究表明CXCL7与中性粒细胞趋化和激活有关。然而,CXCL7在AR特异性病毒应答中的作用尚不清楚,需未来进一步研究。
另一方面,我们发现dsRNA刺激后AR上皮细胞中IL-20、BLNK、CEBPD和LY96特异性下调。IL-20属于IL-10家族,在免疫应答、炎症调节、造血、表皮细胞和角化细胞分化等方面发挥重要作用。IL-20通过Janus激酶(Janus kinase, JAK)-信号转导和转录激活因子(signal transducer and activator of transcription, STAT)途径主要激活STAT3。IL-20可促进多种抗菌肽的产生,增强上皮细胞的屏障功能,并促进白细胞的募集和激活[25]。有研究报道IL-20信号减少会导致炎症和变应原应答增强。Myles等[26]报道IL-20RB信号可抑制皮肤炎症反应,导致IL-1β和IL-17A的产生减少。Wahl等[27]报道体外刺激IL-20RB-/-缺陷的CD8和CD4 T细胞可导致IFN-γ和IL-2分泌显著升高,IL-20RB-/-CD4 T细胞中IL-10分泌减少。他们发现IL-20RB-/-小鼠的一级和二级CD8 T细胞对变应原的反应比IL-20RB+/-小鼠高2~3倍。因此,IL-20可能直接作用于CD8和CD4 T细胞,减弱变应原反应特异性CD8 T细胞的发育。本研究发现在dsRNA刺激后,IL-20在AR上皮细胞中特异性下调;同时,IL-20RB在HC和AR上皮中均下调。然而在无dsRNA刺激时,HC和AR上皮中IL-20、IL-20RA和IL-20RB无差异。我们推测AR中特异性下调的IL-20可能增强变应原应答反应。
BLNK是B细胞抗原受体信号通路的重要组成部分,是B细胞发育所必需的因子。BLNK也在调节B细胞祖细胞向B细胞前体细胞的转化过程中发挥着关键作用[28]。具有CD1dhiCD5+表型并产生IL-10的调节性B细胞称为B10细胞。虽然在BLNK-/-小鼠中存在B10细胞,但其IL10的产生能力在体内和体外都受到了损害。BLNK-/-小鼠也表现出剧烈的接触超敏反应[29]。BLNK还通过介导Jnk/p38的激活促进IgE+B细胞的凋亡。因此,BLNK-Jnk/p38通路缺陷可导致IgE+记忆B细胞和长寿命浆细胞的形成,持续表达IgE从而导致变应性疾病[30]。基于现有的研究证据,我们推测BLNK在AR中特异性的下调可能导致IL-10表达减少和IgE的异常产生,从而促进变应性炎症。
CEBPD作为转录因子参与细胞分化、代谢和免疫应答等多种生物学过程。LY96可增强TLR2介导的细菌应答,并增强TLR4介导的对细菌脂多糖(lipopolysaccharide,LPS)的应答[31-33]。这些基因的下调可能改变效应细胞的功能,并介导下游细胞因子变化以参与AR特异性病毒应答。但其机制尚需进一步研究验证。
基于功能富集分析的方法,我们研究了dsRNA刺激后AR特异的功能和信号通路。其中包括增强的“cell adhesion”和“defense response”,以及受损的“positive regulation of tumor necrosis factor production”。前者可增加炎症细胞浸润,增强先天免疫功能。而对于后者我们的研究发现,在dsRNA刺激后,TNF在AR和HC组均上调,而HC组(logFC:3.037526833)较AR组(logFC:2.674476928)有更高的表达趋势。提示AR上皮在病毒应答时TNF的产生能力下降。有研究报道在病毒性鼻炎中TNF-α的浓度显著高于AR[34]。然而TNF是否与AR的病毒应答有关,以及其详细机制需进一步研究。
在本研究中,我们在基线水平未发现HC和AR之间的DEGs。该实验中体外培养的鼻黏膜上皮细胞平均需要24 d的时间生长到80%的融合状态以接受刺激。因此在离开炎症微环境后,AR表达谱趋于收敛。然而,后续的dsRNA刺激放大了AR的特征基因表达模式。
综上所述,本研究通过比较dsRNA刺激前后AR与HC上皮中DEGs的差异,确定了AR特异性病毒应答相关的转录组特征,并提示上调的hub基因CXCL7以及下调的IL-20、BLNK、CEBPD和LY96可能是鼻病毒诱发AR加重的重要因素。这些结果有助于提示鼻病毒感染导致AR加重的潜在机制,并为下一步的研究提供参考。
