脉冲熔融法测定金属及陶瓷中氧、氮含量的研究进展
2022-03-02姚佳人李任博张重远
张 庸 ,李 瑶 ,姚佳人 ,李任博 ,李 辉 ,张重远
(1.广东腐蚀科学与技术创新研究院,广州 510530;2.中国科学院 金属研究所,沈阳 110016;3.中国航发沈阳黎明航空发动机有限责任公司,沈阳 110043;4.上海材料研究所,上海 200437)
氧、氮对于金属及合金材料的物理及机械性能有较大影响,氧元素性质活泼,通常是一种有害元素[1];氮元素性质较稳定,其作用呈现两面性,缺点是“蓝脆”现象,如造成塑性下降、脆性增加,优点是促进奥氏体化,可部分代替镍[2];氧、氮对于陶瓷材料,特别是氧化陶瓷、氮化陶瓷等的构成有决定作用。同时,氧作为非氧化陶瓷中的杂质,会影响其机械和热稳定性[3]。因此,监控这些材料中的氧、氮含量至关重要。金属中氧分析始于20 世纪初,CUSHMAN[4]采用氢气还原法测定了钢铁中氧的含量,随后,真空熔融法[5]、惰气熔融法[6]相继出现,但分析速率仍有待提高。20世纪60年代中期,脉冲熔融法问世[7-8],这种方法通过对加载有石墨坩埚的水冷电极施加高功率,使样品受热并在短时间内熔融,分析速率明显提高,具有稳定性好、准确度高等优势,目前已成为测定钢铁、钛合金、硬质合金中氧、氮含量的标准方法。相关国家标准有GB/T 11261-2006《钢铁 氧含量的测定 脉冲加热惰气熔融-红外线吸收法》、GB/T 20124-2006《钢铁 氮含量的测定 惰性气体熔融热导法(常规方法)》、GB/T 4698.7-2011《海绵钛、钛及钛合金化学分析方法 氧量、氮量的测定》、GB/T 4324.25-2012《钨化学分析方法 第25部分:氧量的测定 脉冲加热惰气熔融-红外吸收法》、GB/T 4324.26-2012《钨化学分析方法 第26部分:氮量的测定 脉冲加热惰气熔融-热导法和奈氏试剂分光光度法》等。从20世纪90年代起,国内相关综述开始出现[9-13],其内容偏重于国内研究工作,如近期综述[14]和[15]中国外文献仅约占总引文献的30%和10%,且较少涉及以下3方面内容:①设备改进,如设备的关键部分检测、气路系统,关键耗材石墨坩埚;②准确度主要依赖于加标回收,缺少辅助方法的比对;③以条件试验确定最佳参数,反应机理、共存元素干扰等内容论述较少。
国外这方面的研究不乏亮点,比如改进试验设备,以带电粒子活化分析法验证结果,以俄歇电子能谱法解释常规化学清洗不能完全去除样品表面氧的问题[16]。除此之外,俄罗斯巴里舍夫研究院系统研究了钢中不同元素对氧测定的影响,建立了脉冲加热-程序升温法测定氧化夹杂物的理论体系,已成功用于钢铁生产控制[17-18];GRUNER[19]测定了铁、钴、镍等氧化物中的氧含量,指出只配备CO2池的缺陷,可为设备改进提供理论指导。同时,该研究团队还结合X 射线衍射仪等手段探究了氧化钛、氧化锆等氧化物的还原反应机理[20-21],可用于指导上述氧化物的生产。文献[22]详细介绍了俄罗斯巴里舍夫研究院关于氧、氮分析测定方面的工作,引起相关行业从业人员的广泛关注。鉴于此,本工作又汇总了国内外相关文献,结合国内综述,介绍了以下3方面的内容:①设备的相关研究进展,如设备的检测、气路系统的改进,石墨坩埚的类型和差异等;②相关工作的基础理论进展,如反应过程、助熔剂作用以及依据对助熔剂的依赖性对金属分类等;③在具体测试方面,由于常规分析已有汇总[15,22-23],本工作主要综述非常规金属分析和陶瓷分析,如钢中超低氧分析、活性金属及合金分析、氧化陶瓷及氮化陶瓷分析等,重点强调辅助设备的应用、反应机理研究,并进行了国内外比对,以期为相关从业人员提供指导。
1 设备研究进展
氧氮测定仪包括样品加载部分、炉头升降部分、检测系统和气路系统,但具有革新意义的改进主要集中于检测、气路系统。另外,石墨坩埚的研究也不乏亮点,如石墨坩埚的应用进展以及不同类型石墨坩埚带来的测试差异。
1.1 检测、气路系统
氧氮的检测原理[7]为样品在石墨坩埚中熔融,其中氧主要生成CO,而氮以N2释放,混合物通过Cu O,其中CO 被氧化为CO2,用红外法检测,然后去除CO2,用热导检测器检测N2。但该检测理论受到了质疑[19],坩埚中会有部分CO2生成,在催化条件下,CO 转化成CO2的比例与其中CO 含量相关,而CO 不可能全部转化成CO2,这会导致分析结果的测定误差。为了提高测定准确度,有学者曾尝试抑制坩埚中CO2的生成,如吴玲绮等[24]、COLOMBO[25]分别采用坩埚还原法、延迟加热法测定铜、碳化钨中氧含量。GRUNER 课题组系统研究了多种氧化物中CO 及CO2的释放情况,发现CO2释放量不能忽略[19,26],如在研究Mo O3、WO3时,坩埚中以CO2释放的氧分别占总氧量的5.2%,5.7%。GUERRA[27]在催化炉前增加了CO 和CO2(High)池,可同时分析CO 及CO2,修正了只配备CO 池带来的测定误差。KRASOVSKII[28]提出了具体的修正公式,见公式(1)。其中,系数E由标气标定,即在不同功率下,注入CO2标气,以CO2池输出的吸光度为横坐标,CO 池输出的吸光度为纵坐标绘制标准曲线,所得标准曲线斜率的负数为系数E。E不是固定值,例如当含氧量为2.6,13 mg时,E分别为1.01,0.95,当CO2生成量较高时,标准曲线将发生弯曲,不再是一条直线,因此,公式(1)在坩埚中CO2生成量不大于5%(质量分数)时适用。

式中:A,ACO2(High)和ACO分别为总吸光度,CO2(High)池吸光度和CO 池吸光度;E为系数。
除了以上可以用增加红外检测器模块的手段改进外,还可用质谱检测器替代红外检测器和热导检测器,用于钢铁中氧、氮含量的同时测定。UEMURA 等[29]率先提出用飞行时间质谱检测器同时测定氧、氮、氢含量,使气路系统得以简化。某公司[13]研发的脉冲飞行时间质谱仪,已用于钢铁[30]、硬质合金[31]以及中间合金[32]等材料中的氧、氮分析,并编入国家标准GB/T 14265-2017《金属材料中氢、氧、氮、碳和硫分析方法通则》。
在检测N2前需去除CO2,若样品中氧含量过高,例如氧化陶瓷,CO2释放量过大,去除CO2会明显降低气流压力和流量,造成气流不稳,严重影响低氮样品的分析结果,当含氧量为50 000 mg·kg-1,含氮量为5 mg·kg-1时,氮测定值可高达35 mg·kg-1。研究人员尝试采用分流法降低气流中的氧含量,但该方法无法保证待测气体成比例分配至分流气体中,分析精度欠佳。为维持气流稳定,弥补高含量CO2去除对氮测定的影响,有厂家在设备中增加了动态流量补偿器[33],该方法可根据CO2吸收前后的压力变化来调节流量控制阀并补充载气。试验结果显示,加载动态流量补偿器后,即使含氧量高达50 000 mg·kg-1,5 mg·kg-1氮仍可精确测定。
1.2 石墨坩埚
石墨坩埚不仅导电,还可提供碳源。石墨坩埚的尺寸较多,通常分为单坩埚和套坩埚,典型坩埚特点及改善问题见表1。
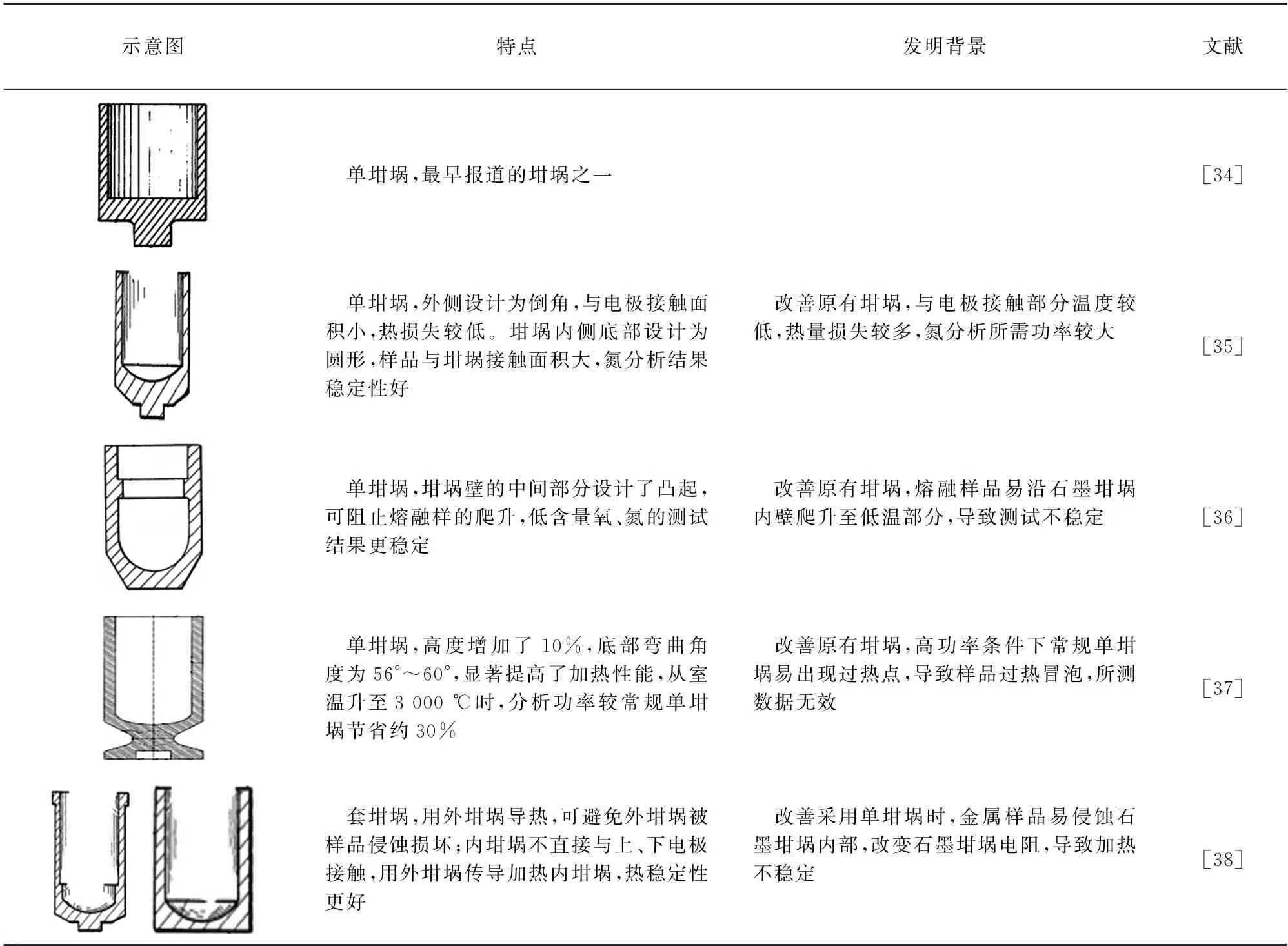
表1 典型坩埚Tab.1 Typical crucible
加热时,坩埚直接接触上、下水冷电极,存在热损失,坩埚不同,热损失也不同,使用单坩埚时此现象比较明显。因此,在加热至相同功率时,坩埚不同,实际加热温度也不同,国外通常采用温度控制模式分析。为使控温准确,部分研究人员选择在测试前对设备温度曲线进行校准[17,39]。但是,坩埚内也存在温度梯度,如分析功率为4.5 kW 时,套坩埚中内坩埚底部温度可达2 500 K,而坩埚盖温度只有约1 400 K[28]。坩埚孔隙率不同,分析结果也可能产生差异。KRASOVSKII等[17]在采用套坩埚进行脉冲熔融-程序升温法分析钢中氧时,采用极孔值分别为18%(多孔坩埚)、6%(稠密坩埚)的内坩埚分析样品,结果显示,多孔坩埚氧分析结果重复性好,而稠密坩埚重复性较差。这种现象与熔融生成的CO 气泡的移除机制有关,气泡在块状熔融物中的氧化物表面形核后,有些气泡会立刻悬浮至熔体表面,有些会黏附至坩埚壁上。对于多孔坩埚,坩埚壁上黏附的气泡多,易离开熔体/石墨接口,释放稳定,而在稠密坩埚中,坩埚壁上黏附的气泡较少,气泡释放不稳定。相对而言,国内通常采用功率控制模式分析,针对坩埚差异的研究多集中于单坩埚和套坩埚,较少关注同类型坩埚温度的差异及理论阐述。王学华等[40]、张长均等[41]经试验确定套坩埚分析精度明显优于单坩埚。前者认为相同功率下,套坩埚温度稍低;而后者认为,在分析功率为1.5~4.0 kW 时,两种坩埚分析温度基本相同,4.0~7.5 kW 时,单坩埚分析温度明显高于套坩埚。谢君等[42]详细比较了单坩埚和套坩埚的具体应用,发现套坩埚的氧空白值较低,氮空白值和单坩埚的相同;针对坩埚损坏以及鼓泡等异常情况,建议及时观察用后的坩埚,以快速发现和剔除异常数据。
2 基础理论
KRASOVSKII等[17]在测定钢中氧、氮时,将反应过程分为三步:①将样品加载至石墨坩埚中,加热至开始熔融;②样品熔融生成碳饱和熔融物,其中氧主要以CO 形式释放,伴有少量CO2生成,氮以N2形式释放;③生成的混合气体经过炉头、试剂以及其他管路等进入检测器检测。其中,后两步是关键步骤,关系到样品中氧是否完全以CO、CO2形式释放,释放的气体是否会被吸附,目标成分能否完全到达检测器。COLOMBO 等[43]发现,Al2O3、SiO2会生成部分亚氧化物Al O、SiO,Au2O3、Ag2O会直接分解生成单质金、银和O2,而Sb2O3、Pb O、In2O3会生成易挥发性氧化物,即并非所有氧均以CO、CO2形式释放。有些样品中的铝、锰、硅等会在高温下蒸发,随后沉积在设备低温部分,易吸收CO,即并非所有气体均可到达检测器。陶瓷材料,特别是氧化物、氮化物等,样品中氧、氮含量高,除存在上述问题外,其气体释放量大,气体体积急剧膨胀,不仅易携带未反应样品颗粒,阻碍反应的持续进行,同时还会形成压力脉冲,扰乱系统气流,干扰测定[28,44]。目前,通常采用添加助熔剂的方法[27]来改变熔融环境,保证后两步反应顺利进行。助熔剂可使氧化物和碳源完全接触,形成低熔点合金,降低熔体的温度和活性金属蒸气压力,增大CO 和N2在熔体上的平衡压力,防止形成黏稠状或固态碳化物[45]。侯桂臣等[46]依据金属对助熔剂的依赖性,将金属分成三大类:第一类为直接分析型,即不需添加助熔剂即可按照上述三个步骤平稳分析的金属,如钢铁、高温合金、铜等,但这些金属的反应机理略有不同,钢铁、高温合金等与石墨坩埚反应性强,熔体表面光滑,铜渗碳性差,熔体呈球状,LÓPEZ等[47]通过Cu-C相图指出,铜和碳形成的物质倾向于包晶型,而非共晶型,溶解碳量通常为mg·kg-1级;第二类为助熔剂依赖型,如钛、锆、铌等纯金属及其合金,这类金属常在第二步熔融时出现问题,需要添加助熔剂来改善熔体状态;第三类为活性金属及合金,此类金属与氧结合力强,较难被还原或金属本身易挥发、易吸附被测气体,后两步反应常出现问题,分析结果会严重偏低且不稳定,所需反应条件比较苛刻。陶瓷材料也可参照上述对金属的分类先分类再进行具体测试。
3 非常规金属分析
3.1 钢中超低氧
钢表面氧的去除直接影响钢中超低氧测定,样品表面粗糙度以优于0.8μm(轮廓算术平均偏差)为宜[48],一般通过化学抛光或电化学抛光等方法获得新鲜表面[49],这也是国内通用的处理方法。但是,借助俄歇电子能谱,YASUHARA 等[16]发现化学处理并不能完全去除钢表面氧,且不同处理方法得到的表面氧化层厚度也不同,如电化学抛光之后氧化层厚度约3 nm,含氧量相当于1 mg·kg-1。随后,采用带电粒子活化分析法(不受样品表面影响)分析氧含量[50-51],结果比脉冲熔融法低1~2 mg·kg-1,进一步说明电化学抛光法并未完全去除表面氧。研究人员尝试采用辉光放电轰击法[52]、两步加热法[53-55]等去除氧化层,其中两步加热法加热条件的选择非常关键,以确保只去除表面氧而基体氧不受影响。UCHIHARA 等[53]发现,钢表面氧化物在750 ℃时开始被还原,900~1 050 ℃内被移除,高于1 050 ℃时样品开始融化。但是,YASUHARA 等[16]发现,钢表面的氧化层被去除后,若再次接触空气,可在1 min内重新生成。针对此问题,UCHIHARA 等[54]改进了样品加载系统,即先在坩埚中于1 000 ℃去除样品表面氧化物后,由磁力棒将样品提取至待加载位置,将炉子升温至2 400 ℃,重新投样分析,无需开闭炉头,解决了样品表面重新氧化的问题。ITO 等[55]利用样品易于重新氧化的特点,先将样品于1 050 ℃氧化,然后从坩埚中取出重新加载,于1 050 ℃加热分析,所得结果即为重新氧化的表面氧含量,结束后从坩埚中再次取出样品并加载,于2 400 ℃加热分析,所得结果为总氧量,总氧量与表面氧含量的差值即为基体氧含量。
钢虽然为直接分析型金属,但YOSHIOKA等[56]认为在分析钢中超低氧时,锡助熔剂的添加必不可少,如1 g样品可选择添加0.75 g锡。UCHIHARA 等[53]、SUZUKI等[57]也认可上述结论,前者认为锡可抑制锰的吸气效应,后者在添加锡铜浴后可准确测定钢中低于10 mg·kg-1的氧。GRIGOROVICH[58]在研究钢中氧化物碳热还原时,发现钢中其他元素,特别是铝、硅等,会明显影响氧的释放。作者也发现,镧、铈等稀土元素会严重影响钢中氧的释放,未加助熔剂时,数据偏低。以上现象表明,共存元素,特别是可产生吸气效应的元素,会影响钢中氧的释放,但现行标准(GB/T 11261-2006等)并未考虑相关影响,希望再次修订时予以补充。
3.2 活性金属及合金分析
活性金属通常包括碱金属、碱土金属、铝以及稀土等,目前国内报道的活性金属主要涉及铝[59-60]、镁[61]、钒铝[62-63]、铬铝、钼铝[64]、硅钙[65]、镧铈合金[66]等,对其试验参数进行汇总,结果见表2。刁伟涛[59]指出:带盖石墨坩埚可抑制Al O 及铝蒸气的挥发,进而使AlO 被CO 还原;以Sn-Cu作助熔剂,可控制熔体中的碳含量,避免固态碳化物析出;同时锡蒸气压大,可抑制铝挥发;此外,该研究还借助气体状态方程,验证了氧提取反应已完全。表中其他研究偏重于试验参数探讨,理论研究较少,甚至出现了无法解释的截然相反的试验现象,如一些研究认为镍助熔剂可抑制铝的挥发[62-63],促进Al2O3中氧的还原,而其他研究则认为镍箔、镍篮会抑制、阻碍Al2O3中氧的释放[64]。同时,由于缺少相应标准样品,这些研究多采用异标校正,如高纯物质、钛合金标准样品、钢标准样品等,通过测定这些物质回收率或者加标回收率来保证准确度,而异标校正是否会导致测量差异,这些研究并未深入探讨,并且辅助方法应用较少,除文献[62]和文献[66],其他未见采用辅助方法验证。

表2 国内文献中活性金属及合金测定的典型试验参数Tab.2 Typical test parameters for the determination of active metals and alloys in domestic literatures
国外相关报道在理论研究以及辅助方法的使用等方面不乏亮点。INOUE 等[67]在测定Fe-Al合金中氧时,绘制了Al、Al O、Al2O 以及Ni中Al吉布斯自由能(以下简称自由能)随温度变化的热力学相图(图1,其中g代表气体,M 为Al2O3)。结果显示,Al O、Al、Al2O 及Ni中Al自由能依次降低,这是由于镍助熔剂的添加会抑制Al2O 以及铝挥发,这与文献[62-63]现象一致。

图1 AlO、Al、Al2 O 及Ni中Al的热力学相图Fig.1 Thermodynamic phase diagram of AlO,Al2 O,Al,and Al in Ni
同时,该研究团队还确认Al2O3颗粒在金属浴的溶解为速率控制过程,熔体应保持较好的流动性,不应过于黏稠。而文献[64]采用镍篮分析时,助熔剂和样品的质量比高达100∶1,镍具有中等容碳性,熔体中碳过多,与其中高达60%的钼反应生成过饱和的碳化钼[68],致使熔体过于黏稠,Al2O3颗粒难以被还原,生成的气体也难以脱离熔体,测试结果偏低。综上,镍可抑制铝测试时Al2O 及铝的挥发,但镍不应过量,否则易导致熔体黏稠,阻碍气体生成及释放。TSUGE 等[39]在测定镁中氧时,强调由于镁的强挥发性,需将基体镁与Mg O 分离,即先将样品与锡在900 ℃下共熔,随后升温至2 000 ℃,此时,镁被蒸发至坩埚边缘,Mg O 与锡则残留于坩埚底部,加热还原MgO,用红外检测器检测,测试过程见图2。X 射线荧光光谱法结果表明,镁与Mg O已完全分离;与带电粒子活化分析法进行比对,结果显示,在分析低含量氧时,两种方法结果一致,分析高含量氧时,带电粒子活化分析法结果明显偏高,这是由样品加工过程中引入的氢氧化物不能被脉冲熔融法检测而被带电粒子活化分析法检测所致。综上可知,深入的理论分析可从根本上解释异常现象,而带电粒子活化分析法等辅助方法能更好地验证方法的准确度。

图2 镁中氧测试过程示例Fig.2 Schematic of the measurement procedure of oxygen in Mg
4 陶瓷分析
4.1 氧化物、氮化物
陶瓷材料测试难点主要集中于氧化物、氮化物,特别是易还原及挥发氧化物、氮化物陶瓷。这类物质反应快速,气体释放量大,例如含20%~45%氧的25 mg氧化物陶瓷会释放3~8 mL CO(相对室温)。高温下,气体急剧膨胀且易携带未反应颗粒进入气流,扰动系统压力,阻碍反应平稳进行,甚至导致坩埚顶部形成圆顶。针对这个问题,国外报道较多,通常采用助熔剂[69]以及延迟加热[70]等方式来避免气体快速释放和保证反应平稳进行。其中延迟加热通常采用程序升温模式,该模式下加热速率慢、气体释放较平缓。TSUGE等[69]指出金属浴的促进能力按照由小到大排序依次为Ti、Sn、Cu-Ni、Fe,但单元浴所得测定值明显低于理论值,而双元浴Sn-Ni、Sn-Fe由于可抑制圆顶形成,所得测定值与理论值相吻合。DUBOK 等[3]采用熔融铜将坩埚内表面覆盖,以阻止样品的挥发。BECK 等[70]在分析陶瓷材料中Fe2O3、Cr2O3中氧时,所得数值偏低,认为是由于样品与助熔剂镍同时熔融,导致大量CO 生成,气流被扰乱,氧不能被全部检测。但GRUNER[19]不认同此观点,此课题组在系统研究氧化物氧释放行为时,发现Bi2O3、Fe2O、MoO3、NiO 及WO3等氧化物均有不可忽略的CO2生成,而老型号设备无法测定直接生成的CO2,导致检测结果偏低。
氮化物陶瓷分析,起初主要集中于Si3N4[71-73]。ADELHELM 等[71]指出,分析氮化物陶瓷时,其中的含硅氧化物易被还原生成的SiO 干扰,这与COLOMBO[43]观点一致,可采用过量镍抑制SiO 生成。WÖRNER 等[72]、KITAYAMA 等[73]则分别采用Ni-Cu 浴、石墨粉作助熔剂。WATANABE等[74]则借助化学前处理测定了氮化硅的表面氧。TANAKA 等[75]、WATANABE 等[76]则采用压样机压制镍囊,提高了检测效率。在准确度保证方面,GRUNER 等[77]指出:TiN(wN=22.62%)、Si3N4(wN=39.94%)等常用标准样品的均匀性相对较差,且含有大量氧化物杂质;ε-TaN 为线状化合物,均匀性好,湿度较大时仍旧稳定,可通过钽板或钽箔制备标准样品。但GRUNER 等[44]却对异标校正提出异议,即校准应是以相同含量的氮为准还是以信号强度为准,如10 mg氮化硼与22 mg氮化硅含氮量相同,但信号强度分别为400~500 和750~800,而以这两种标准物质校正后得到的含氮量分别为55.6%,55.2%,至于哪种校正模式最佳,该团队并未深入探讨。
国内关于氧化物、氮化物的报道较少。朱跃进等[78]在分析烧结氮化硅时指出:热力学因素并非是检测过程的主导因素;动力学分析结果表明,烧结块不易熔化和溶解,溶解及传质过程均为速率控制过程;2 450 K 下会生成固态SiC 硬壳,阻止融熔反应进一步进行,改变浴料组成可改善样品的熔化和分散状态,如当采用五元浴作助熔剂时,结果与化学法一致。
4.2 氧化物反应原理
样品熔融时,氧化物中部分氧以CO2形式释放。KRASOVSKII[28]认为CO2量主要取决于坩埚内反应条件;氧化物的分析步骤与钢中氧[17]略有不同,其中氧首先以不同形式进入气体,Cu O、NiO、CoO、Fe2O3等主要以歧化形式反应,MoO3、WO3升华,Sn O2、GeO2则是介于前两者之间的形式释放,这与COLOMBO[25]观点类似,即氧化物反应形式除氧化还原外,还包括歧化、分解反应等;熔融时,坩埚内会形成高氧化势能区(PO2=10-5~10-1MPa),而碳表面的氧势能则急剧降低(PO2=10-18~10-20MPa),氧相对过量,形成CO2;随后CO2转化为CO,而转化速率主要取决于CO2在坩埚热区的存在时间以及扩散至碳表面的速率,当加上坩埚盖时,CO2在坩埚中的停留时间延长,CO2的生成量将不会超过4%~5%,采用WO3、Mo O3、Fe2O3、Cu O 和Sn O2等高纯物质进行验证,除部分Sn O2数据有偏差外,其他高纯物质均无明显偏差。
GRUNER 团队[20-21]利用脉冲熔融法检测,同时借助质谱仪以及电子显微镜等,系统研究了Zr O2、TiO2、SiO2的反应机理,指出Zr O2、TiO2反应步骤类似,都需经过三步反应,以TiO2为例,其碳热还原过程见图3(a),即:①TiO2被CO 还原生成固态中间产物TinO2n-1;②TinO2n-1进一步被还原生成碳氧化物,碳的结合和氧的损失速率并不等同;③碳氧化物继续被还原,生成相应的碳化物。由此可见,碳化物的生成是以氧化物为基准,质量传输由CO/CO2实现。SiO2的碳热还原过程[20]与上述步骤明显不同[图3(b)],且与文献[43]和文献[71]强调应抑制SiO 生成不同。因为反应首先生成中间产物SiO,此中间产物为气态,然后以碳为基准生成碳化物,质量传输主要是由SiO 实现,这与现场生产及热分析结果一致。

图3 TiO2 和SiO2 的碳热还原过程的反应机理Fig.3 Reaction mechanism of the carbothermal reduction of TiO2 and SiO2
5 结语
脉冲熔融法测定氧、氮含量已广泛用于金属及陶瓷粉末生产质量控制,但目前国内工作仍存在对设备关注较少,依赖经验分析,准确度还主要依赖加标回收,缺少理论分析以及其他试验方法验证等缺点,而国外这方面可借鉴的亮点比较多。为了提升国内对于金属及陶瓷中氧、氮分析能力,可从如下建议入手:①加强设备原理的研究,特别是坩埚部分,需关注不同坩埚的差异,如功率相同时,坩埚尺寸不同,分析温度也会有差异;②利用合理有效的辅助方法验证分析结果的有效性,如对钢中超低氧分析,若无俄歇电子能谱,则难以发现化学处理表面氧尚存在的不足,准确度试验主要依赖于加标回收,无法避免不同物质反应差异带来的影响;③加强理论研究,特别是反应机理的研究,这不仅可以从根本上解释加标回收、异标校正差异性,也可以更好地选择试验条件。
