易错PCR定向进化技术提高蒙古黄芪病程相关蛋白AmPR-10核酸酶活性的研究
2022-02-28张育敏闫海洋胡丽丽
王 珏,吴 娜,张育敏,闫海洋,胡丽丽
(山西中医药大学基础医学院,山西 晋中 030600)
病程相关蛋白(pathogenesis-related protein,PR)是植物在遭受病原菌侵害或外界环境压力下产生的一种抗逆的、有利于自身生长发育的蛋白质,分为17个家族,其中PR-10类蛋白具有核酸酶活性[1],被认为与降解RNA类病毒有关[2]。蒙古黄芪病程相关蛋白-10(Astragalusmembranaceuspathogenesis-related protein-10,AmPR-10)由158个氨基酸残基组成,分子大小约为16.8 kDa。目前已有研究证明天然提取的和重组的AmPR-10都具有核酸酶活性,但是活力均偏低[3-4],不利于更深入的研究及大范围的应用。
酶分子的定向进化是由美国科学家Arnold F H于1993年首先提出,技术理念是通过模拟自然进化机制,对酶基因进行体外修饰,产生基因多样性,利用高通量筛选技术,获得具有预期特性的修饰酶[5]。定向进化可利用多种手段诱导目的基因碱基发生增添、缺失、替换等突变,代表性的方法为易错PCR[6]。易错PCR定向进化技术不需要对蛋白质结构有充分的了解,目前已在获取高活性的几丁质酶A[7]、γ谷氨酰转移酶[8]、β-1,4-葡聚糖内切酶[9]等方面得到了应用。
作者所在课题组[4]前期利用大肠杆菌表达体系构建了AmPR-10的2个重组子:pET30a-AmPR-10及pET30a-skp-AmPR-10,其中skp是来源于大肠杆菌的分子伴侣,由161个氨基酸构成。外源诱导表达结果显示分子伴侣skp修饰后,AmPR-10表达量显著提高。因此,作者选择分子伴侣skp修饰的AmPR-10全基因skp-AmPR-10为研究对象,通过易错PCR定向进化技术筛选核酸酶活性提高的AmPR-10突变体,为进一步探索AmPR-10的作用机制提供理论依据,为其它同源蛋白的基因定向进化提供参考。
1 实验
1.1 材料、试剂与仪器
重组子pET30a-skp-AmPR-10由山西中医药大学基础医学院克隆合成。
感受态细胞E.coliBL21、质粒提取试剂盒、超级感受态制备试剂盒、蛋白纯化试剂盒、96孔细菌培养板、2×Taq PCR Master Mix,生工生物工程(上海)有限公司;限制性内切酶NcoⅠ、XhoⅠ、T4 DNA连接酶,赛默飞世尔科技(中国)有限公司。
TC-XP-G型PCR仪,杭州博日科技有限公司;TGL23M型高速冷冻离心机,湖南湘立科学仪器有限公司;BSD-400型振荡培养箱,上海博讯实业有限公司;01A037型超声破碎仪,宁波新芝生物科技股份有限公司;WD-9413B型凝胶成像分析仪,北京六一生物科技有限公司。
1.2 方法
1.2.1 AmPR-10基因定向进化模板的选择
参照文献[4]进行重组子pET30a-AmPR-10及pET30a-skp-AmPR-10构建过程中skp及AmPR-10的扩增、引物的设计、重组子的克隆、目的蛋白的诱导表达等。通过比较重组子pET30a-AmPR-10及pET30a-skp-AmPR-10对目的基因的表达情况,发现后者可溶性表达量更高,因此,确定pET30a-skp-AmPR-10为目的基因定向进化的野生型。
1.2.2 目的基因skp-AmPR-10的易错PCR
以重组子pET30a-skp-AmPR-10的质粒为易错PCR模板,在PCR体系中添加不同浓度(0 mmol·L-1、0.1 mmol·L-1、0.2 mmol·L-1、0.3 mmol·L-1、0.4 mmol·L-1、0.5 mmol·L-1)的Mn2+以确定合适的突变率。以skp-AmPR-10作为目的基因设计上下游引物,上游引物:5′-CTAGCCATGGTGAAAAAGTGGTTATTAGCT-3′(划线部分为限制性内切酶NcoⅠ的酶切位点);下游引物:5′-CCGCTCGAGCTTGTATTCAGGATTGGCCA-3′(划线部分为限制性内切酶XhoⅠ的酶切位点)。PCR体系为:2×Taq PCR Master Mix 25 μL,质粒模板1 μL,上游引物1 μL,下游引物1 μL,氯化锰溶液1 μL,加ddH2O补足50 μL。
1.2.3 超级感受态的制备及突变体文库的构建
将获得的含有一定突变率的目的基因经过酶切回收后与载体pET30a连接,将连接产物加入到提前取出冰上融化的E.coliBL21感受态细胞中,按超级感受态制备试剂盒操作说明制备转化效率更高的超级感受态。将复活后的菌液涂布于含有卡那霉素的LB固体培养基上,37 ℃培养30 min后,将培养皿倒置过夜培养,构建转化子数量较高的突变体文库。
1.2.4 单克隆的诱导表达
将固体培养基上的菌落逐个挑到96孔细菌培养板中(每孔含200 μL已添加抗生素的LB培养基),于37 ℃、200 r·min-1培养12~16 h;然后以2%的接种量将各孔中的菌液转接到新的96深孔板中(每孔含500 μL已添加抗生素的LB培养基),加入IPTG进行诱导表达,剩余菌液与甘油以7∶3的比例混匀作为母板于-80 ℃冰冻保存;离心收集诱导后的菌体,于-80 ℃过夜冻存;每孔加入200 μL的RIPA细胞裂解液,于37 ℃放置1~2 h使菌体裂解;于4 ℃、4 000 r·min-1离心20 min,取上清,测定目的蛋白活性(以OD260值表征)。
1.2.5 高通量筛选方法的构建
以酵母tRNA为底物,构建AmPR-10核酸酶活性检测的高通量筛选方法。具体如下:配制2 mg·mL-1的酵母tRNA溶液,于96孔紫外酶标板中每孔加入50 μL酵母tRNA溶液与50 μL 菌体裂解后的蛋白液(以野生型蛋白液为对照),37 ℃反应30 min;反应结束后通过酶标仪测定反应体系的OD260值(测3组平行数据,取平均值),与对照组进行比较,筛选正向突变体。
1.2.6 突变体的复筛
高通量筛选方法建立的体系是微量的,在一定程度上存在误差,常用于目的基因定向进化的初筛。为进一步测定突变体的表达及活性,将初筛得到的突变体进行扩大培养,超声破碎菌体获得目的蛋白;通过SDS-PAGE检测蛋白表达情况,纯化突变体蛋白;通过考马斯亮蓝法测定蛋白浓度。以酵母tRNA为底物,扩大反应体系,测定蛋白比活[10]。
2 结果与讨论
2.1 易错PCR Mn2+浓度的确定
分子伴侣skp由483个碱基组成(不包括终止密码子),AmPR-10由477个碱基组成(包括终止密码子),所以全基因skp-AmPR-10的扩增条带大小为960个碱基。在PCR体系中,分别加入不同浓度Mn2+的目的基因扩增产物的鉴定结果如图1所示。

M.DNA marker 1~6,Mn2+浓度(mmol·L-1):0、0.1、0.2、0.3、0.4、0.5
由图1可以看出,加入不同浓度Mn2+的目的基因扩增产物均在960 bp处显示明显条带;Mn2+可以降低PCR过程中Taq聚合酶的保真性,随着Mn2+浓度的增加,突变率升高,条带亮度也随之降低。因此,既要保证有一定的突变率,又要保证PCR产物量足够,选择0.3 mmol·L-1Mn2+进行突变体基因的扩增。
2.2 高通量筛选AmPR-10突变体结果
第一轮定向进化共筛选单克隆890个,然后通过高通量筛选方法测定96孔细菌培养板中所有单克隆突变体核酸酶活性(OD260值),并计算扣除缓冲液样品对照平均值后的OD260值,结果见表1。
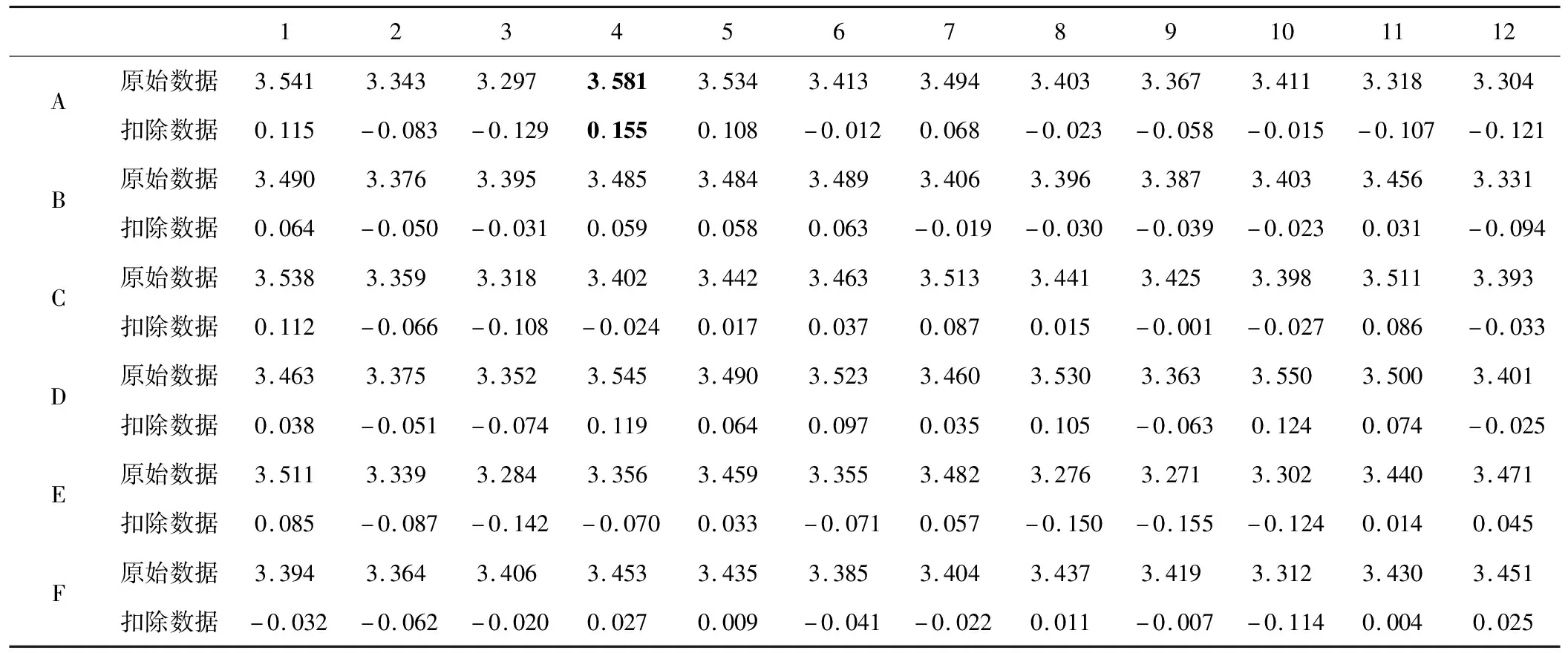
表1 突变体核酸酶活性检测原始数据及扣除缓冲液样品对照平均值后的OD260值
由表1可知,位于A4位置的突变体的核酸酶活性明显提高;缓冲液样品对照E1、F1、G1、H1的原始数据平均值为3.426,野生型样品对照A1、B1、C1、D1的扣除数据平均值为0.082,而突变体A4的扣除数据(0.155)最大。表明突变体A4的核酸酶活性可能较野生型有所提高,需要进行扩大培养,对其进行复筛。
2.3 突变体A4核酸酶活性的复筛和基因测序
野生型蛋白及突变体A4蛋白的表达及纯化结果如图2所示。
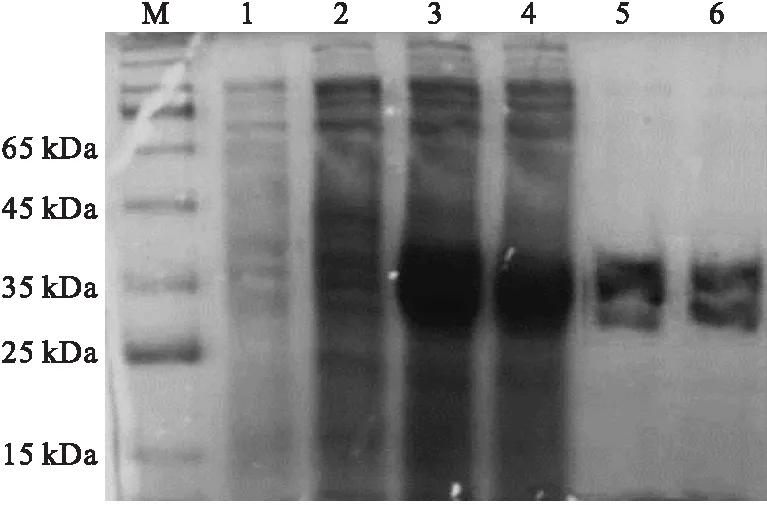
M.蛋白质marker 1,3,5分别为野生型蛋白未诱导、诱导及纯化后的鉴定结果 2,4,6分别为突变体A4蛋白未诱导、诱导及纯化后的鉴定结果
skp与AmPR-10融合表达后,蛋白大小约为34 kDa。由图2可以看出,突变体A4蛋白诱导和纯化后的SDS-PAGE图谱均在约34 kDa处出现明显条带(泳道4、6),符合预期;并且与野生型(泳道3、5)相比,突变体A4的表达量并没有明显减少,说明基因突变没有影响目的蛋白的可溶性表达。
分别对野生型蛋白及突变体A4蛋白进行纯化,测定蛋白浓度;以酵母tRNA为底物,测定两者的比活。结果发现,野生型蛋白的比活为1.1 U·mg-1,突变体A4蛋白的比活为1.6 U·mg-1,较野生型提高45%。
野生型蛋白及突变体A4蛋白的基因测序结果如图3所示。
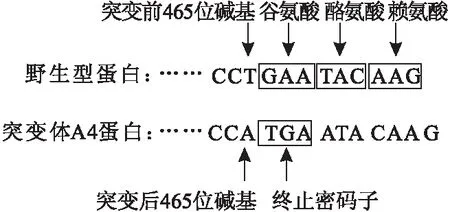
图3 野生型蛋白及突变体A4蛋白的基因测序结果
由图3可以看出,与野生型蛋白相比,突变体A4蛋白在465位处多了一个碱基A,使得第153个氨基酸密码子由CCT突变为CCA,但均编码脯氨酸,并导致后续基因排列的移码突变。很有意思的现象是,由于移码的出现,使得原来第154个氨基酸密码子变为TGA终止密码子,因此,突变体A4在蛋白编码过程中会提前终止,与野生型蛋白相比,缺少末端3个氨基酸,依次为:谷氨酸(Glu)、酪氨酸(Tyr)、赖氨酸(Lys)。而且实验证明这3个氨基酸的缺失并没有影响目的蛋白的表达和活性,这一结果对探讨AmPR-10核酸酶活性机制具有重要参考意义。
通过SWISS-MODE建模,获得野生型蛋白和突变体A4蛋白的空间结构,使用Chimera软件对其进行初步分析,结果如图4所示。
由图4a可以看出,突变体A4蛋白缺失的3个氨基酸位于AmPR-10空间结构C末端的一段长α螺旋上(在SWISS-MODE建模中,最后一位赖氨酸没有显示),已有研究[10]报道,这段区域对于AmPR-10展现核酸酶活性而言,保守性不强,可以作为基因定向进化的参考位点,而突变体A4蛋白的测序结果也对这一理性分析提供新的证据;另一方面,C末端的α螺旋结构犹如手柄状,对AmPR-10的空腔形成遮蔽作用,而空腔具有配体结合活性,在目的蛋白抗逆过程中发挥作用[11]。因此突变体A4在缺失了3个氨基酸后,α螺旋的长度缩短,进而对空腔的遮蔽作用减小,有利于蛋白与配体或底物的结合,这或许是突变体A4核酸酶活性提高的原因。
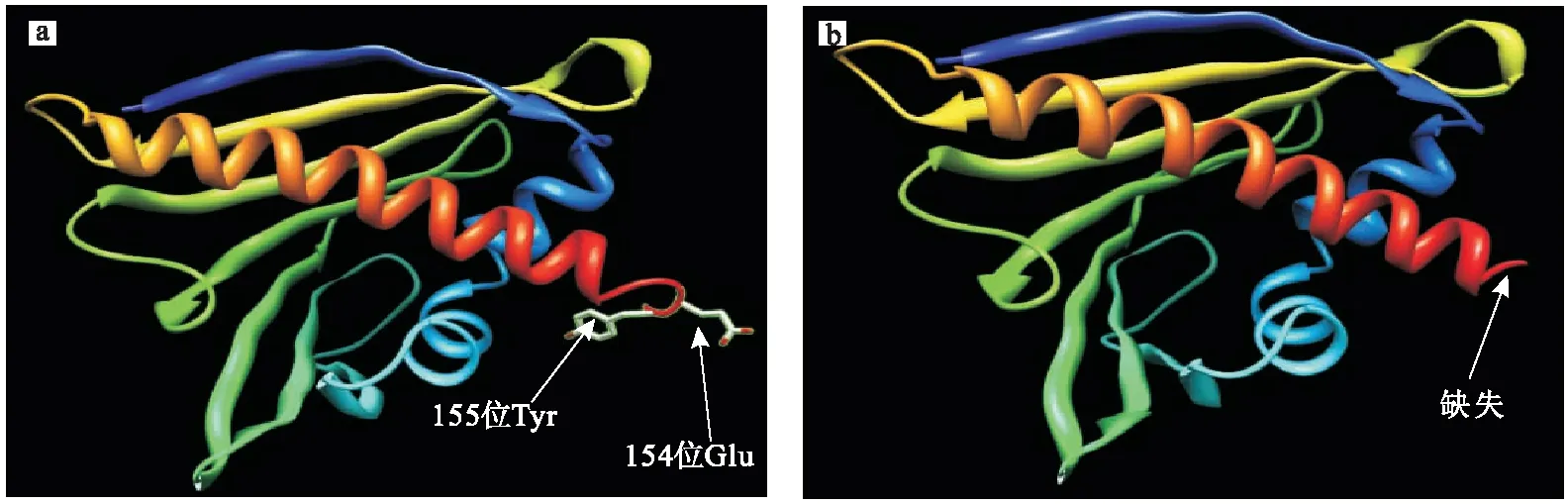
图4 野生型蛋白(a)和突变体A4蛋白(b)的空间结构示意图
2.4 讨论
(1)选择分子伴侣skp修饰的AmPR-10全基因skp-AmPR-10为研究对象,这一策略使分子伴侣skp分担了定向进化过程中的突变压力,可以避免仅以AmPR-10为目的基因时,进化范围(基因片段)较小而造成易死突变的现象,并且在一定程度上推迟进化平台期的出现,增加突变的多样性。研究方法利于正向突变体的筛选。
(2)以分子伴侣skp与AmPR-10串联的全基因为模板(skp位于AmPR-10上游),进行了一轮易错PCR随机突变定向进化,得到了1个活性提高的突变体A4,其活性并没有达到理想状态,仅比野生型提高45%,其空间结构的C末端缺失了3个氨基酸(谷氨酸、酪氨酸、赖氨酸),但并未影响到目的蛋白的表达和活性。因此在后续研究中,可考虑以突变体A4为基础,进行第二轮定向进化,筛选活性进一步提高的突变体;或结合分子伴侣的作用,针对skp进行改造,通过优化目的蛋白的折叠和表达,实现核酸酶活性提高的目的。筛选核酸酶活性提高的AmPR-10突变体,是一项持续而重要的工作,在此过程中,可逐步挖掘影响其活性的关键位点或区域,从而得到高活性的AmPR-10,这对优化蒙古黄芪育种、降低种植过程中病原体侵袭造成的损失具有重要意义。
3 结论
以分子伴侣skp修饰的AmPR-10全基因为模板,通过易错PCR定向进化技术构建突变体文库,以酵母tRNA为底物建立高通量筛选方法,获得了核酸酶活性提高的AmPR-10突变体。突变体A4的核酸酶活性比野生型提高45%;基因序列在465位增加了1个碱基A,C末端缺失了3个氨基酸,从而减小了其C末端α螺旋对空腔的遮蔽作用,有利于目的蛋白与配体或底物的结合。该研究结果对AmPR-10核酸酶活性机制的探讨具有重要意义,为抗病抗逆黄芪植株的培育奠定了理论基础。
