沉积物活化氧气和过氧化氢产生羟自由基降解三氯乙烯的比较研究
2022-02-27滕晓宇郑云松蔡其正卢钰茜袁松虎
滕晓宇,郑云松,蔡其正,卢钰茜,张 鹏*,袁松虎,2
1.中国地质大学(武汉),生物地质与环境地质国家重点实验室,湖北 武汉 430078
2.中国地质大学(武汉),长江流域环境水科学湖北省重点实验室,湖北 武汉 430078
原位化学氧化(ISCO)是一种修复污染土壤或地下水的常用技术[1].通过向污染土壤/含水层注入化学氧化剂,利用物理/化学方法将氧化剂活化为高氧化活性中间体,从而使有机污染物被氧化转化为H2O、CO2等无害物质[2-5].过氧化氢(H2O2)是目前ISCO 修复工程中广泛应用的氧化剂[6],当H2O2被注入地下环境后,其与外源添加或含水层中的Fe(Ⅱ)等还原态物质发生(类)芬顿反应,生成大量羟自由基(·OH,标准氧化还原电位为2.8 V),从而实现对污染物的氧化去除[7-10].虽然目前许多以H2O2作为氧化剂的研究都取得了较好的修复效果,但是普遍存在氧化剂迁移距离短、稳定性差、寿命短、氧化剂有效利用率低等问题[11-13].根本原因是,H2O2进入含水层后,除与Fe(Ⅱ)反应生成·OH 外,还可能发生不产生·OH 的过氧化氢酶和过氧化物酶型反应[14],使H2O2分解转化为H2O 或O2.此外,H2O2成本较高,并且在储存、运输等方面存在一定风险[15-16].
近年来研究发现,地下环境受O2扰动时,沉积物中含量丰富的还原性组分〔主要为Fe(Ⅱ)和有机质〕可以活化O2产生·OH,从而有效地氧化转化四环素、苯酚和三氯乙烯(TCE)等多种污染物[3,17-19].其中,·OH 的产生机制可简要地归结为Haber-Weiss 机制,即Fe(Ⅱ)首先活化O2产生·O2−和H2O2,进一步地,Fe(Ⅱ)与H2O2反应生成·OH[17].因此,从科学原理的角度分析,相比于Fe(Ⅱ)活化H2O2产生·OH,Fe(Ⅱ)活化O2转化为·OH 仅需要消耗更多的Fe(Ⅱ).地下环境中Fe(Ⅱ)储量丰富[18],具备将O2充分还原为·OH 进而降解污染物的可能性,但目前关于沉积物活化O2和H2O2产生·OH 去除污染物之间的定量差异尚不清楚.
鉴于此,该研究拟通过室内静态试验体系,探究不同类型沉积物活化O2、H2O2产生·OH 的产量和氧化剂转化率的差异,并采用TCE 作为代表性污染物来评估两种氧化剂体系产生的·OH 对污染物的去除作用,通过定量对比不同条件下,沉积物活化O2、H2O2产生·OH 的能力和氧化剂转化效率,以及去除污染物的差异,以期为评估O2在ISCO 中的应用潜力提供理论支持.
1 材料与方法
1.1 材料
苯甲酸钠(BA,99.5%)、对羟基苯甲酸(p-HBA,99%)、乙二胺四乙酸二钠(EDTA,99.0%)、三聚磷酸盐(TPP,98.0%)、TCE(99.6%)等均购买自国药集团.试验中超纯水(18.2 MΩ·cm)由Heal Force NW 超纯水仪制得,无氧水由超纯水经高纯N2吹脱1 h 并在厌氧手套箱(96% N2和4% H2,COY,美国)中搅拌12 h制得.
试验用沉积物由手持土壤取样钻机(SD-1,澳大利亚)分别在某河岸带和某化工厂两个场地采集.沉积物岩芯取出地面后,现场快速用保鲜膜和锡箔纸包裹,然后装入真空袋密封抽真空,转运到实验室保存在−20 ℃冰柜.沉积物质地、总Fe(Ⅱ)含量、不同形态Fe(Ⅱ)含量、含水率和可溶性有机碳含量等指标见表1.
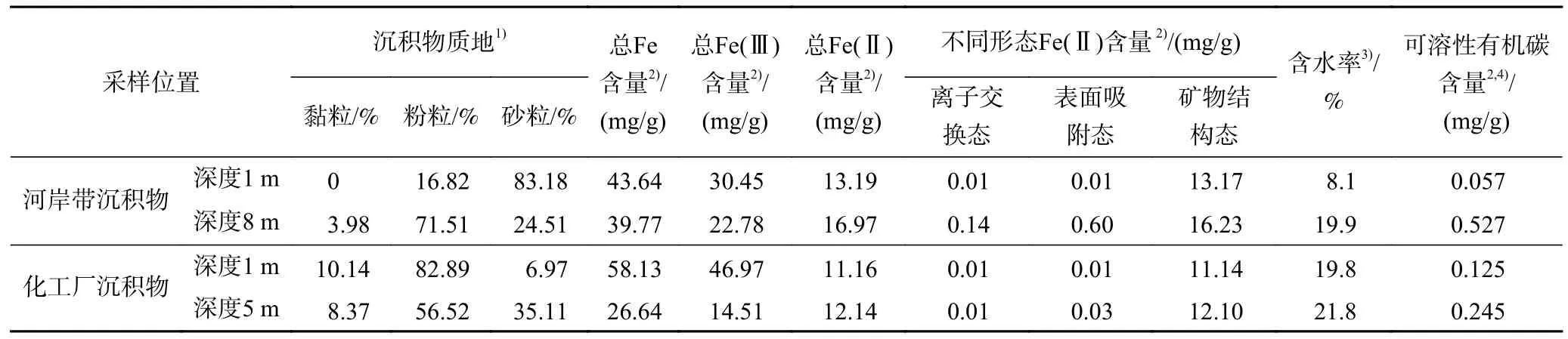
表1 沉积物基本性质Table 1 Basic properties of sediment
1.2 试验过程
所有试验均在体积为120 mL 的厌氧瓶中进行.为了避免光照,厌氧瓶外包裹锡纸.试验开始前,取4 g湿沉积物与80 mL 无氧水在手套箱中混合,然后向上述悬浊液中加入20 mmol/L BA,使用稀H2SO4和NaOH 调节初始pH 至7.0,最后用丁基胶塞和铝盖密封.反应开始前,将厌氧瓶取出手套箱,利用注射器迅速从胶塞处注入一定体积O2(纯度为80%)或H2O2储备液,使体系中初始O2和H2O2浓度分别为4.6 mmol/L(该文所述的O2浓度均为假定体系中O2完全溶解于水相的浓度)和5.0 mmol/L,然后将反应器置于恒温摇床(25 ℃,220 r/min)上开始反应.其他试验均在此基础上调节部分参数,其中探究配体强化·OH产生和TCE 去除的试验中,对应体系添加EDTA 或TPP 的浓度均为1.0 mmol/L,探究污染物降解的试验中,采用12 μmol/L TCE 代替20 mmol/L BA.此外,为探究配体促进沉积物活化O2产生·OH 的机制,该研究单独测定了溶解态Fe(Ⅱ)-配体体系中·OH 的产生规律.以上所有试验均至少进行2 次,结果以平均值±标准偏差表示.
1.3 分析方法
于不同时间点从厌氧瓶中取出1 mL 悬浊液,过0.22μm 滤膜后,测量p-HBA、H2O2及TCE 的浓度.为了淬灭取样后·OH 与BA 的反应,取0.5 mL 甲醇与0.5 mL 滤液混合.通过高效液相色谱仪(16C HPLC,Shimadzu,Japan)测定p-HBA 浓度[18],然后将其乘以系数5.87 换算出累积·OH的浓度[20].H2O2和溶解态Fe(Ⅱ)的浓度分别采用硫酸钛显色法[21]和邻菲罗啉显色法测定[22].水相DO 浓度和气相氧分压均利用非侵入式便携光纤氧分析仪(Fibox 4,Pre-Sens GmbH,Germany)测定,氧气贴片粘置于厌氧瓶内壁距瓶底1/3 瓶身处,将水相DO与气相氧分压之和记为体系瞬时O2总浓度.每片氧气贴片上有相应的集成条形码,第一次使用前扫描后方可实现传感器的识别和校准.滤液中TCE 经正己烷萃取后,采用气相色谱仪(GC2014 C,Shimadzu,Japan)测定[23].
沉积物基本性质测定方法:沉积物经10%H2O2和10%HCl 去除有机质和无机碳后,加入0.05mol/L(NaPO3)6使沉积物分散,然后采用激光粒度分析仪(MS 3000,马尔文仪器公司)测定沉积物颗粒粒度分布[24].采用化学分步提取法测试不同形态Fe(Ⅱ)的含量,具体为1mol/L CaCl2、1mol/L NaH2PO4和3.6mol/L H2SO4+1mol/L HF 分别提取离子交换态、表面吸附态和矿物结构态Fe(Ⅱ)[18],三者之和即为沉积物固相Fe(Ⅱ)总含量.根据烘干前后沉积物质量差与湿质量之比得到沉积物含水率.沉积物经0.1mol/L NaOH提取后,通过总有机碳分析仪(TOC-L,Shimadzu,Japan)测定,得到沉积物可溶性有机碳含量.
1.4 氧化剂转化率计算方法
根据Haber-Weiss 机理[25],沉积物中Fe(Ⅱ)活化O2产生·OH 的过程如式(1)~(3)所示,其中式(3)也是沉积物中Fe(Ⅱ)活化H2O2产生·OH 的途径,式(4)为H2O2无效分解产生H2O 和O2.
由式(1)~(3)可知,每生成1 mmol/L·OH 需要对应消耗1 mmol/L O2或1 mmol/L H2O2,因此氧化剂转化为·OH 的效率计算方法如下:

式中:Y·OH为 氧化剂转化为·OH 的效率,%;C·OH为累积产生·OH 的浓度,μmol/L; ΔC为ΔCO2或ΔCH2O2,分别表示累积消耗O2或H2O2的浓度,μmol/L.
类似地,根据式(4),每生成1 mmol/L O2需要消耗2 mmol/L H2O2,故H2O2分解为O2的效率计算方法如下:
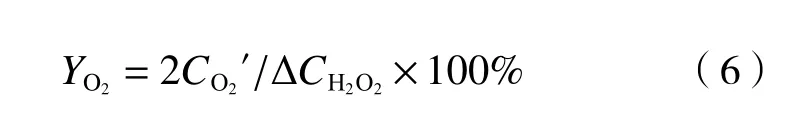
式中:YO2为O2的转化率,%;CO2′为H2O2体系中累积产生O2的浓度,μmol/L.
2 结果与讨论
2.1 沉积物活化O2 和H2O2 产生·OH 的差异比较
该研究选用河岸带和化工厂两个场地不同深度沉积物作为代表性沉积物,分别进行活化O2和H2O2产·OH 的对比试验.结果显示,向河岸带地下1 m 和8 m 以及化工厂地下1 m 和5 m 沉积物悬浊液(50 g/L)中分别注入4.6 mmol/L O2后,在180 min 内产生的累积·OH 浓度分别为0.5、7.1、1.0、13.8 μmol/L(见图1).相比于埋深为1 m 的沉积物,河岸带地下8 m与化工厂地下5 m 沉积物均可以更加有效地活化O2产生·OH,可能原因是后者含有更多的Fe(Ⅱ)(见表1),更加有利于·OH 的产生.已有研究[26]也报道了类似结果,即沉积物中Fe(Ⅱ)含量越高,曝氧氧化后产生·OH 的浓度也越高.反应结束后,不同类型沉积物体系中残余O2的浓度均高于3.9 mmol/L,说明O2是足量的.但是·OH 的产生随时间的延长呈先快后慢的趋势,可能原因是,表面活性Fe(Ⅱ)在初期阶段快速消耗,而沉积物内部电子来不及向表面转移[27-28].根据式(5)计算,河岸带地下1 m 和8 m 以及化工厂地下1 m 和5 m 沉积物活化O2转化为·OH 的效率(即·OH 产率)分别为0.1%、1.1%、0.2%、3.0%(见表2).
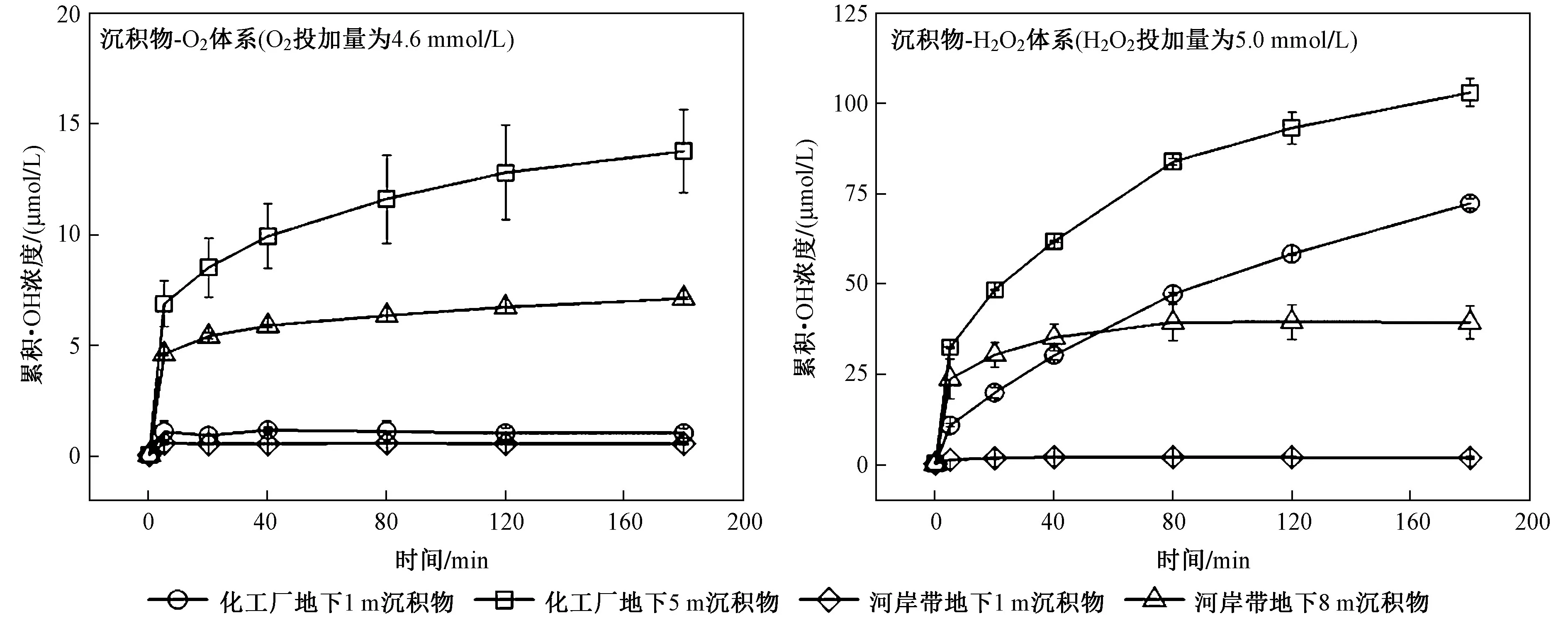
图1 沉积物-O2/H2O2 体系中累积·OH 浓度随时间的变化规律Fig.1 Variations of cumulative ·OH concentration versus time in sediment-O2/H2O2 systems
当向河岸带地下1 m 和8 m 沉积物以及化工厂地下1 m 和5 m 沉积物悬浊液(50 g/L)中分别注入5.0 mmol/L H2O2后,在180 min 内产生的累积·OH 浓度分别达到1.7、39.1、72.1、102.8 μmol/L,远高于沉积物-O2体系(见图1).H2O2与河岸带地下8 m 和化工厂地下5 m 沉积物反应后,残余H2O2浓度分别仅为0.01 和1.03 mmol/L,说明H2O2的反应活性较O2高.但是根据式(5)计算,河岸带地下1 m 和8 m以及化工厂地下1 m 和5 m 沉积物活化H2O2产生·OH 的效率(即·OH 产率)分别为0.03%、0.8%、0.9%、2.4%(见表2),与O2转化为·OH 的效率接近,但是远低于100%.究其原因,可能是部分H2O2无效分解为O2和H2O〔见式(4)〕,如化工厂地下5 m 沉积物-H2O2体系中H2O2分解为O2的比例(即O2产率)高达92.0%(见表2).上述结果表明,虽然在数小时内沉积物-O2体系产生·OH 的累积浓度小于沉积物-H2O2体系,但是·OH 产率却与H2O2处于相近水平.
2.2 氧化剂投加量对·OH 产量和产率的影响
为进一步探究不同氧化剂投加量对·OH 产量及其产率的影响,该研究以河岸带地下8 m 沉积物为代表,分析O2投加量为0、2.3、4.6、7.0 mmol/L(假定O2完全溶解于水相的浓度)和H2O2投加量为0、0.5、1.0、5.0、10.0 mmol/L 条件下·OH 的产生规律.随着O2投加量从0 mmol/L 增至2.3 mmol/L,180 min 内累积·OH 浓度从0.5 μmol/L 提升到6.7 μmol/L,而O2投加量进一步从2.3 mmol/L 增至7.0 mmol/L,·OH 产量则由6.7 μmol/L 缓慢地增至7.5 μmol/L(见图2),·OH产率由1.5%降至0.8%(见表2),相当于O2投加量每增加1 mmol/L,·OH 产率却降低了10%.这可能归因于沉积物中活性Fe(Ⅱ)含量有限以及其他还原性物质对O2消耗的影响.
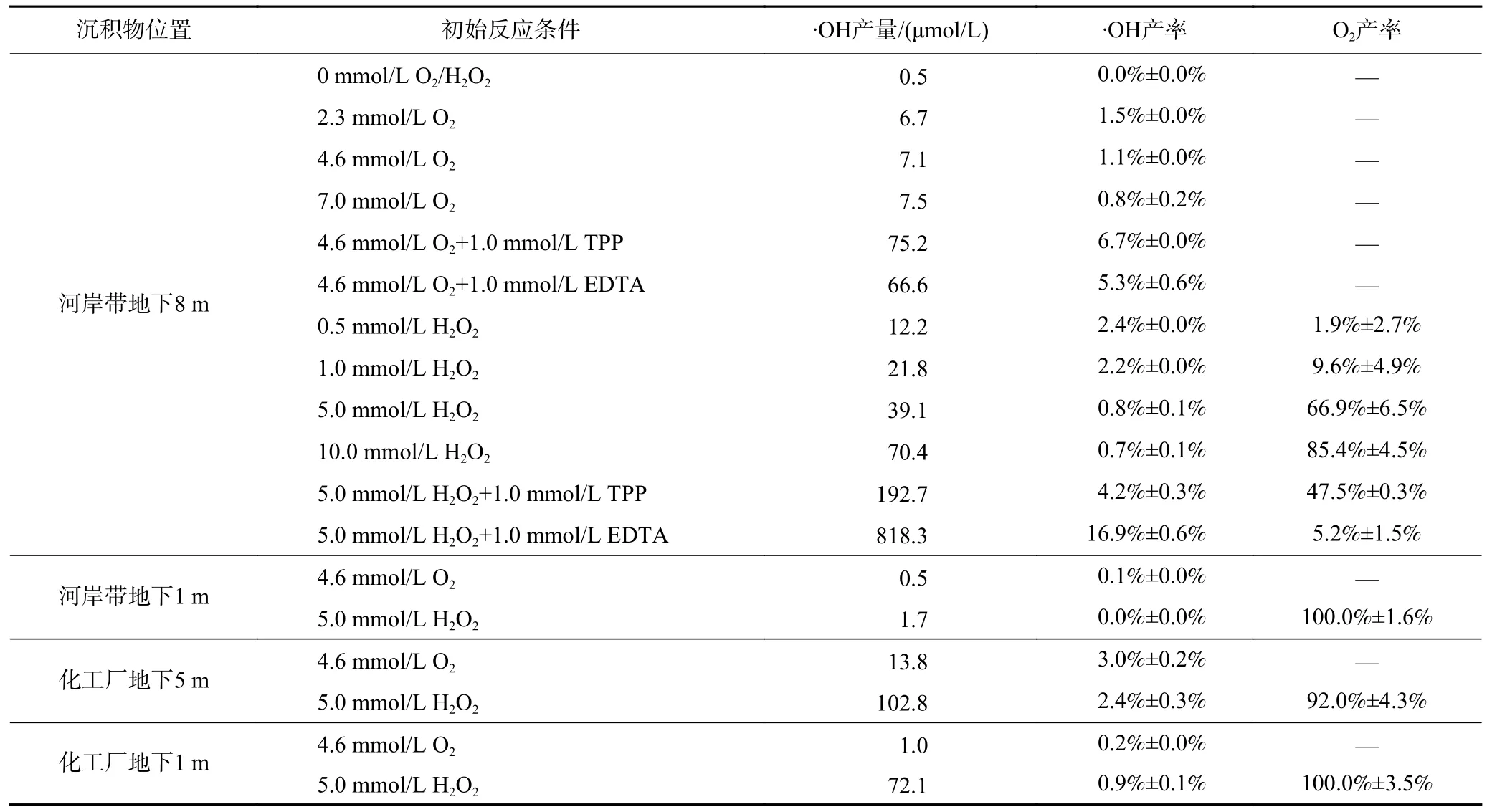
表2 沉积物-O2/H2O2 体系中·OH 产量和·OH 产率以及H2O2 分解为O2 的效率Table 2 The yield and efficiency of ·OH in sediment-O2/H2O2 systems and the efficiency of O2 production from H2O2 decomposition
已有研究[15-16]报道,沉积物中表面吸附态Fe(Ⅱ)和矿物结构态Fe(Ⅱ)是O2被活化产生·OH 主要的电子供体,且沉积物中Fe(Ⅱ)的反应活性高低表现为离子可交换态Fe(Ⅱ)>表面吸附态Fe(Ⅱ)>矿物结构态Fe(Ⅱ).在河岸带地下8 m 沉积物悬浊液(50 g/L)中离子可交换态Fe(Ⅱ)、表面吸附态Fe(Ⅱ)、矿物结构态Fe(Ⅱ)初始浓度分别为0.12、0.54、14.5 mmol/L.测量结果显示,氧化后总Fe(Ⅱ)浓度减少了0.3~0.75 mmol/L,可能主要归结为离子可交换态Fe(Ⅱ)和表面吸附态Fe(Ⅱ)的快速氧化.若表面吸附态Fe(Ⅱ)全部参与反应〔见式(1)~(3)〕,需消耗0.18 mmol/L O2,远小于初始O2投加量.因此,即使O2投加量增加,吸附态Fe(Ⅱ)与O2反应的·OH 产量也不变,表观上体现为·OH 产率下降.虽然矿物结构态Fe(Ⅱ)也可以贡献·OH 的产生,但其与O2的反应速率很低,故短时间内与O2反应的·OH 产量有限.因此,随着O2投加量的增加,·OH 产量无明显提升.实际反应过程中O2消耗量为0.53~1.03 mmol/L(见图2),大于吸附态Fe(Ⅱ)对O2的消耗量,这可能是沉积物中其他形态Fe(Ⅱ)物种〔如离子可交换态Fe(Ⅱ)〕、有机质和生物酶等还原性组分与O2反应以及好氧微生物呼吸,造成了氧化剂损失.
随着H2O2投加量从0.5 mmol/L 增至10 mmol/L,180 min 内累积·OH 浓度由12.2 μmol/L 增至70.4 μmol/L(见图2),对应的·OH 产率由2.4%降至0.7%,同时H2O2分解生成O2的效率(即O2产率)从1.9%显著增至85.4%(见表2).因此,H2O2转化为·OH 的产率随H2O2投加量的增加而降低可以归结为高浓度H2O2无效分解为O2.从H2O2的消耗趋势可看出,当其投加量不超过0.5 mmol/L 时,H2O2在5 min 内即被完全分解,累积·OH 浓度在5 min 达到最大值,随后保持不变;当投加量大于0.5 mmol/L 时,H2O2在前5 min 分解速度很快,之后下降,累积·OH 浓度随反应时间的延长而增加(见图2).加入H2O2反应180 min 后,沉积物中总Fe(Ⅱ)浓度减少了0.1~1.1 mmol/L.因此,与O2类似,H2O2在初期阶段的快速消耗也可能与表面活性Fe(Ⅱ)有关.当表面活性Fe(Ⅱ)被消耗后,沉积物中Fe(Ⅲ)矿物、含锰矿物、有机质、污染物及其他还原性物质等组分均会显著地贡献H2O2的分解[14],从而导致大量H2O2被转化为O2和H2O.综上,增加氧化剂浓度有助于提升·OH 产量,但对应·OH 产率却呈下降趋势.

图2 不同氧化剂投加量对沉积物-O2/H2O2 体系中·OH 产生和氧化剂消耗的影响Fig.2 Effects of oxidant dosage on ·OH production and oxidant consumption in sediment-O2/H2O2 systems
2.3 配体强化下O2 和H2O2 体系中·OH 产率对比
为提高氧化剂的有效利用率,增加·OH 产量,该研究分别将有机配体EDTA 与无机配体TPP 加入沉积物-O2/H2O2体系.结果显示,配体的加入有效地促进了两种氧化剂体系中·OH 的产生.例如:在沉积物-O2体系中,未加入配体时180 min 内·OH 产量为7.1 μmol/L,加入1.0 mmol/L EDTA 和TPP 后,·OH 产量分别提高到66.6 和75.2 μmol/L(见 图3).在沉积物-H2O2体系中,未加入配体时180 min 内·OH 产量为39.1 μmol/L,而加入1.0 mmol/L EDTA 和TPP 后,·OH产量分别提高到818.3 和192.7 μmol/L(见图3).与未加入任何配体时相比,加入EDTA 后,沉积物-O2体系的·OH 产率提升了381.8%,而沉积物-H2O2体系提升了2 012.5%(见表2);加入TPP 后,沉积物-O2体系的·OH 产率提升了509.1%,而沉积物-H2O2体系提升了425.0%(见表2).加入配体后,·OH 产量和产率的增加,可能归结为配体的络合作用降低了Fe(Ⅱ)/Fe(Ⅲ)的电极电位,使其具有更强的还原活性[29-30],从而提高了O2和H2O2与Fe(Ⅱ)反应产生·OH 的速度和效率.
由于配体的加入主要影响了溶解态铁的形态[18],因此进一步研究配体对水相Fe(Ⅱ)活化O2或H2O2产生·OH 的影响.结果显示,在与悬浊液体系溶解态Fe(Ⅱ)浓度(0.25 mmol/L)相等或者更高(0.50 mmol/L)的纯水相Fe(Ⅱ)与O2或H2O2体系中,也得到了与沉积物悬浊液体系相似的·OH 产生规律(见图3),说明均相Fe(Ⅱ)氧化可能是配体促进沉积物悬浊液体系产·OH 的重要机制.

图3 配体对沉积物-O2/H2O2 体系和Fe(Ⅱ)-O2/H2O2 体系中·OH 产生的影响Fig.3 Effect of ligand on ·OH production in sediment-O2/H2O2 and Fe(Ⅱ)-O2/H2O2 systems
2.4 沉积物-O2/H2O2 体系中TCE 降解效果对比
未加入配体时,在河岸带地下8 m 沉积物-O2体系中,反应180 min 内TCE(初始浓度为12 μmol/L)的去除率为15.5%(见图4).厌氧空白对照组中TCE含量维持不变,说明沉积物的吸附、还原和微生物作用以及TCE 的挥发等因素对TCE 去除可以忽略.当向上述反应体系加入1.0 mmol/L EDTA 后,TCE 的去除率增至27.0%,而加入1.0 mmol/L TPP 后,5 min内TCE 的去除率达到67.4%,随后逐渐增至100%(见图4).试验中EDTA 的总浓度降低了60 μmol/L,而TPP 的浓度基本保持不变,因此两种配体存在条件下,TCE 去除率的差异可以归结为EDTA 与TCE 竞争消耗·OH[23].
相比之下,在未加入配体条件下,沉积物-H2O2体系中,180 min 内TCE(初始浓度为12 μmol/L)的去除率仅为7.7%,小于沉积物-O2体系.当向反应体系加入1.0 mmol/L EDTA 或TPP 后,在5 min 内TCE 的去除率达到100%,随后保持不变(见图4).上述结果表明,加入配体可以有效促进两种氧化剂体系中TCE 的降解.相比于EDTA,TPP 对TCE 降解的促进作用更强,究其原因:①EDTA 会与TCE 竞争消耗·OH,从而在一定程度上不利于TCE 的氧化降解;②已有研究[18]表明,相比于EDTA,TPP 对Fe(Ⅱ)的络合能力更弱,因此加入TPP 后体系中溶解态Fe(Ⅱ)的 含量较沉积物-EDTA 体系要低.溶解态Fe(Ⅱ)和·OH 的二级反应速率常数为5×108(mol/L)−1s−1,接近TCE 与·OH 的二级反应速率常数〔4×109(mol/L)−1s−1〕[31].因此,相比于EDTA 体系,TPP 溶解固相Fe(Ⅱ)对TCE 降解所产生的竞争作用可能更弱.因此,与·OH 竞争作用弱且化学性质更为稳定的TPP可能是强化沉积物活化O2产生·OH 氧化降解有机污染物的优选配体.笔者也注意到,加入配体对TCE 降解率的促进程度明显高于·OH 产率,这可能与TCE初始浓度较低有关.该研究中TCE 初始浓度为12 μmol/L,在不考虑其他物种与TCE 竞争消耗·OH 的情况下,只需要12 μmol/L·OH 就可以将TCE 完全降解.加入配体后,·OH 产量大幅提高,远大于TCE 的初始浓度,有可能实现了TCE 的完全降解.
2.5 应用O2 作为ISCO 修复工程的氧化剂的启示与展望
该研究发现,含Fe(Ⅱ)的沉积物活化O2产生·OH 的效率略高于H2O2.在特定配体强化下,O2转化为·OH 的效率相比于H2O2转化为·OH 的效率提升更为显著,并且均能够实现TCE 的完全氧化去除.上述结果说明,O2具有作为氧化剂应用于ISCO 修复工程的潜力.然而受沉积物表面活性Fe(Ⅱ)含量的限制,短时间内O2与沉积物反应产生的·OH 浓度较低,远不及H2O2.已有研究表明,当黏土矿物表面Fe(Ⅱ)被氧化后,结构内部电子会逐渐转移到矿物表面,使表面吸附态和结构边缘态Fe(Ⅲ)还原再生为Fe(Ⅱ),从而继续与O2反应,贡献·OH 的产生[32].由于硅酸盐结构态Fe(Ⅱ)是沉积物中主要的铁形态(见表1),因此可以推测O2作为氧化剂有望在较长时间尺度上持续产生·OH.
虽然O2氧化反应速率适中、稳定性相对较好、有效利用效率高,并且O2廉价易得,但如何将O2靶向供给至污染含水层是应用O2作为氧化剂在ISCO修复中需要克服的难题.目前常见地下水原位供氧的方法有地下水曝气、投加释氧化合物和电解水等[33-38],但不同供氧方法的经济成本、供氧效率以及技术要求不同,因此后续研究需要结合污染场地水文地球化学特征来综合评估供氧方式.此外,该研究证实添加配体可以有效促进污染物的氧化去除,因此同时向污染含水层注入O2和配体可能会取得较好的修复效果.
3 结论
a)O2作为氧化剂的有效利用率和H2O2处于相近水平,但是在短时间内O2被沉积物活化产生的·OH 产量较H2O2低.
b)两种氧化剂体系中,随着氧化剂投加量的增加,·OH 产量也随之增加,但是氧化剂分解为·OH 的效率降低.
c)加入配体可有效提高沉积物-O2/H2O2体系中·OH 的产量和氧化剂转化效率,但是相比于沉积物-H2O2体系,配体对沉积物-O2体系的调控作用更强.
