石墨烯基二氧化碳电化学还原催化剂的研究进展
2022-02-26杜亚东孟祥桐汪珍赵鑫邱介山
杜亚东,孟祥桐,3,*,汪珍,赵鑫,邱介山,2,*
1北京化工大学化学工程学院,有机无机复合材料国家重点实验室,化工资源有效利用国家重点实验室,北京 100029
2大连理工大学化工学院,辽宁省能源材料化工重点实验室,精细化工国家重点实验室,辽宁 大连 116024
3中国科学院山西煤炭化学研究所,中国科学院炭材料重点实验室,太原 030001
1 引言
作为一种温室气体,CO2的减排和转化利用是国内外关注的热点和难点之一。中国陆地区域大气中的CO2浓度已超过400 ppm (1 ppm = 1 × 10-6,体积分数),高于政府间气候变化委员会(IPCC)所规定的安全上限(350 ppm)。要实现2060年碳中和之目标,需要研究CO2的高效转化途径。目前,CO2的处理方式主要有两种:CO2的捕集与储存以及将CO2转化为化学品1-3。CO2的转化方式主要包括:催化加氢4,5,光催化6-8,电催化9-15,生物转化16等。CO2的电催化还原(Electrocatalytic CO2Reduction Reaction,ECR)是近年来的研究热点,其突出优势包括:操作环境为常温常压,可降低设备局限性并提高操作安全性;可调控施加电位和催化剂改变产物选择性;可利用间歇性的清洁能源(如太阳能、风能以及潮汐能等)驱动ECR过程17。目前,ECR面临一些局限与挑战,主要包括:CO2是一种热力学稳定的线性分子,需要较高的还原电位方能将CO2分子转变为CO2·-中间体,该步骤与电催化剂的性质密切相关,是ECR所面临的主要挑战和瓶颈之一18;该反应涉及多电子和质子转移,产物多样。反应体系的常规电解液为水系,常伴随竞争性的析氢反应(HER),进而影响ECR产物的法拉第效率19;在反应过程中,中间产物(如甲酸或者甲酸盐)、副产物甚至是电解液中的杂质离子(如Fe3+或者Zn2+)会毒害或阻塞催化剂的活性位点,这将严重影响ECR催化剂的稳定性20。在使役工况下,ECR催化剂的结构重排(如金属单原子和金属团簇的团聚、官能团的还原、介微观形貌坍塌等)以及活性组分的价态、组成的变化(如金属还原导致的空位消失、高价金属被还原为金属单质等)等是影响自身催化活性和稳定性的另一关键原因。因此,开发高性能、高选择性、高稳定的电催化剂,对于实现CO2高效转化、实现我国2060年碳中和目标是十分必要的。
ECR是一个多步反应过程,根据外加电位以及催化剂的不同,涉及的转移电子数也有所差异,通常包括2e-、4e-、6e-、8e-、12e-、14e-以及18e-等反应过程,导致产物多样性21。ECR的产物多达10余种,主要为C1(CO、HCOO-/HCOOH、CH3OH、HCHO、CH4等)、C2(C2H4、C2H5OH、H2C2O4/C2O42-、C2H6等)和C2+(如C3H7OH、CH3COCH3)等产物22,23。表1是在水系电解液中不同产物所对应的阴极半反应式(vs.Standard Hydrogen Electrode,SHE)。与之相对,阳极则发生析氧反应(OER)。对于OER过程而言,其理论起始电位较高(达1.23 Vvs.RHE(Reversible Hydrogen Electrode))且反应动力学缓慢,能耗占据ECR体系约90%。因此,在阳极选取低起始电位(vs.RHE)的氧化反应过程取代OER是降低电解能耗、提高能量效率的有效方式之一15。施剑林等以甲醇氧化反应取代了阳极的OER,甲醇的理论起始电位为0.103 V,远小于OER的起始电位,是一个较为理想的替代反应。除甲醇以外,甘油氧化(0.25 Vvs.RHE)、尿素氧化(0.37 Vvs.RHE)以及甲烷氧化(0.63 Vvs.RHE)等氧化反应的理论起始电位均明显小于OER的起始电位,是OER的理想替代反应15,24,25。各ECR还原产物对应的反应机理取决于催化剂性质,图1列举了部分C1和C2产物(包括CO、CH4、HCOO-/HCOOH、C2H4和C2H5OH)的反应机理18,26-28。近年来,随原位表征技术的发展,对于ECR反应机理的研究在不断深化和完善。

图1 CO2电还原产生部分C1和C2产物反应机理Fig.1 Reaction mechanism of partial C1 and C2 products from CO2 electroreduction.

表1 CO2不同电化学还原产物电势表Table 1 Standard redox potentials for ECR products.
ECR催化剂性能的评价参数主要为法拉第效率(Faradaic Efficiency,FE)、过电位(η)、电流密度(Jtotal,Jproduct)以及稳定性(Stability)等。其中η代表电催化过程中施加的电位与产物产生的热力学平衡电位之差,与催化剂本体性质有关,直接显示催化剂的催化能力并可反映ECR过程的能量效率;电流密度是反应活性的体现,可分为总电流密度与单一产物电流密度。稳定性是指催化剂在催化反应过程中保持活性、产物选择性、抗毒性等性质的能力,也指催化剂的使用寿命,是评价催化剂性能优劣的重要指标之一。这些参数的优化是设计高性能ECR催化剂的原则之一。图2为目前ECR测试体系中较常用的H型电解池示意图,两室由离子交换膜(如常用的Nafion 115、Nafion 117等)分隔。催化剂作为工作电极置于阴极室,将通入其中的CO2气体催化还原。尽管H型电解池的使用较为普遍,其存在较大的缺陷:一是CO2在水系电解液中的溶解度较低(常温常压下溶解度仅为33 mmol·L-1),导致电流密度较小;二是较大的欧姆电阻(阴阳极之间存在几厘米的间隔)以及离子交换膜的存在等导致较低的能量转化效率。而流通池和膜电极电解池则有效改善了上述问题。这两种类型的电解池可以实现气体反应物与液体电解液持续通入反应体系。此外,搭配使用气体扩散电极会在电解池中形成气液固三相界面,令CO2以气体形式发生还原反应而不需溶于电解液中再扩散至电极表面还原,极大地增强了传质动力学,从而表现出较大的电流密度,更具有商业化前景29。流通池与膜电极电解池相比而言,两者最大的不同是流通池阴阳极与离子交换膜之间仍然有电解液的存在,因而与H型电解池类似存在较大的欧姆电阻。此外,由于流通池使用气体扩散电极,故可采用利于ECR过程的强碱性溶液作为电解液以加速CO2还原。而在膜电极电解池中,阴阳两极之间只有膜,并且充当固态电解质,因此减小了电极间的距离,表现出较小的串联电阻和较高的能量转换效率29。与电解池所匹配的电解液在ECR过程中也发挥着重要作用,电解液的差异对于产物种类以及分布等均有明显影响,Resasco等研究发现电解液阳离子的改变影响产物选择性30。目前,常用电解液仍以无机电解液为主,包括用于H型电解池的碳酸氢盐(KHCO3、NaHCO3等)以及用于流通池的氢氧化物(KOH、NaOH等)。离子液体、甲醇和乙腈等有机电解液也被应用于ECR过程,这些有机电解液在某些方面比无机水系电解液更有利于ECR过程,如具有较大的CO2溶解度、能够在一定程度上抑制HER、提供了低温CO2电还原的可能(0 °C以下进行);但此类有机电解液存在缺点:因缺少质子来源而导致产物多为CO、成本较高不利于大规模应用。因而设计、筛选合适的电解液也是发展ECR的重要一环,如Wang等提出一种“Water-in-salt”电解液,将高浓度的盐与水混合,通过强溶剂化作用使游离的水转化为溶剂化水,从而有效抑制水的活性,进而抑制了HER反应31。除此之外,通过优化电极/电解液界面性质可调控界面上的电荷传输动力学、反应物/反应中间体的吸附行为,进一步提高电催化活性。如孙晓明等在电极表面修饰季铵盐阳离子促进CO2的传输与还原,同时抑制了HER32;Hwang等则在多孔锌电极表面包覆还原氧化石墨烯来改变反应物的传质过程,提高ECR性能的同时,对于HER也有明显的抑制作用33。

图2 电解反应池及催化剂分类Fig.2 The reaction cell and the classification of graphene-based catalysts for ECR.
目前,ECR的催化剂多为金属催化剂,包括金属的单质形态、化合形态、不同金属的合金形态及金属的复合材料等。根据产物的不同,金属催化剂划分为:主产C1产物的金(Au)、银(Ag)、锌(Zn)、钯(Pd)、铅(Pb)、锡(Sn)、铋(Bi)、铟(In)等;主产C2及C2+产物的铜(Cu)基催化剂34-44。金属基催化剂种类繁多,不同程度上存在一些限制或缺陷:高活性催化剂多为贵金属,成本较高,不利于ECR的商业化应用;通常有较高的过电位及较差的稳定性45,46。与金属材料相比,碳材料具有来源广泛、成本低、导电性良好及稳定性高等优势;作为ECR催化剂时,可显著降低反应过电位,有助于提高能量效率。目前,用于ECR催化剂的碳材料主要有碳纳米管47,48、碳纤维49、石墨烯50-53、富勒烯54、多孔碳和衍生碳(金属有机框架材料(MOF)、生物质)等55-59。其中,石墨烯作为碳材料的基本构筑单元,可通过卷曲形成0D富勒烯及1D碳纳米管,或堆叠形成3D石墨等(图3),其具有高理论比表面积(2600 m2·g-1)、独特的电子结构和优异的机械强度60,61。石墨烯基催化剂材料(无掺杂石墨烯、杂原子掺杂石墨烯、石墨烯基单原子催化剂以及石墨烯复合材料,如图3所示)的设计构筑是目前ECR领域的研究热点之一。本文将对石墨烯基ECR催化剂的研究进展进行系统总结(表2),包括石墨烯基电催化材料的构筑技术方法(如湿化学法、原位掺杂、静电诱导组装等)、理化性质表征(X射线吸收谱、原位拉曼/红外等)、构效关系解耦(如石墨烯表面官能团、点/线缺陷、掺杂原子构型、金属单原子种类、材料表界面性质等与ECR性能的关系)、反应机制诠释(活性位点、电子转移数、反应路径等)。另外,以石墨烯为载体和导电基质负载活性组分,能防止活性组分的团聚,可设计并构筑高活性和高稳定性的ECR催化剂。本文的最后,对石墨烯基材料在ECR中的前景进行展望,分析面临的机遇和挑战。本文旨在为发展石墨烯基CO2高效电催化转化的技术提供参考。
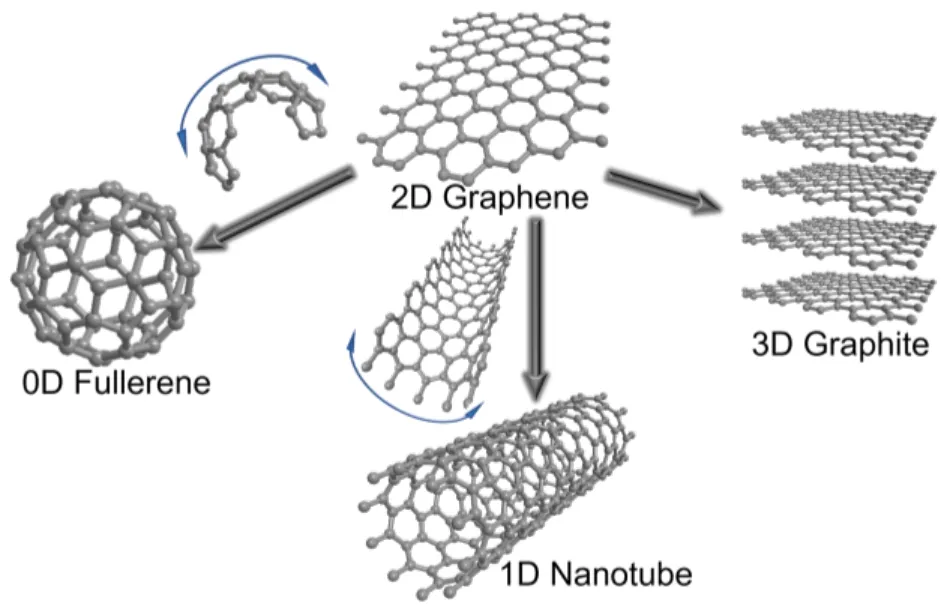
图3 不同维度石墨化碳材料的构筑单元Fig.3 Building blocks of graphitized materials with different dimensionalities.

表2 用于ECR的石墨烯基催化剂Table 2 Summary of graphene-based catalysts for ECR.
continued Table 2
2 石墨烯基催化剂
石墨烯虽有极佳的理化性质,但其电还原CO2活性并不突出,原因是石墨烯基面中的中性碳原子较难活化CO2分子,同时对反应中间体的吸附能力较弱90。石墨烯边缘位置的碳原子相较于基面碳原子不稳定,通常表现出较高的催化活性,因此需要对石墨烯进行剪裁以暴露更多边缘位点或对其基面进行活化(如引入碳空位、掺杂杂原子、修饰活性组分等)来提高石墨烯整体的电催化性能91,92。下面将具体介绍活化石墨烯的具体策略。
2.1 无掺杂石墨烯
在石墨烯制备与加工过程中不可避免的会形成结构缺陷,缺陷包括点缺陷和线缺陷。点缺陷可分为单空位缺陷、多空位缺陷及拓扑缺陷(如Stone-Wales缺陷);线缺陷可分为位错缺陷及边缘缺陷等。这些缺陷对于石墨烯的电催化性能有显著影响91,93。以拓扑缺陷为例,在不改变石墨烯本身sp2杂化情况下,对六元碳环进行局部重构会形成五元环、七元环或八元环等拓扑构型94。通过在碳材料中掺杂杂原子,再经过热处理移除杂原子是制备缺陷碳材料的常规手段50,62。
陈亮等利用NH3处理策略从富N多孔碳骨架中移除吡啶N和吡咯N,制备出具有高密度拓扑缺陷的多孔碳;作为ECR催化剂,富缺陷多孔碳对于CO的选择性高达95.2%62。郑耿峰等利用类似方法,制备出可将CO2还原为CO的缺陷石墨烯(DG),在-0.6 Vvs.RHE电位下选择性达84% (图4)。发现利用移除掺杂N原子方法形成的拓扑碳缺陷不仅提高了电化学活性表面积及对CO2的吸附能力,同时大大降低了电荷转移电阻(Rct),表明材料导电性及界面电子转移能力有所提高50。
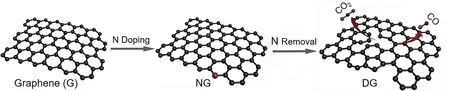
图4 缺陷石墨烯(DG)合成示意图50Fig.4 Schematic illustration of the synthesis of DG50.
除在石墨烯中制造结构缺陷以外,石墨烯的表面官能化也是常用的活化方式。徐维林等用硝酸处理石墨烯纳米盘(GNDs),在其表面接枝含氧官能团(―COOH、―OH、C―O―C和C≡O等)并探讨了其ECR活性51,发现含氧官能团的存在可催化CO2还原为甲酸盐,法拉第效率可达到86%(-0.68 Vvs.RHE)。发现GNDs表面含氧官能团种类和含量影响ECR产物选择性,其中,羧基(―COOH)含量与ECR性能呈正相关,是影响石墨烯活性的主要因素。密度泛函理论(Density Functional Theory,DFT)计算表明含氧官能团附近的带电碳原子是ECR过程中真正的活性中心。同时该催化剂表面的羧基基团可以通过电氧化方法再生,从而提高了催化剂的耐久性。
2.2 杂原子掺杂石墨烯
除对石墨烯进行官能化及制造本征碳缺陷外,向石墨烯中引入杂原子(N、P、B、S、Si、Se等)是提高石墨烯电催化性能的另一个简单易行的方式。掺杂原子主要是与碳原子电负性和共价半径不同的非金属原子,图5为常见的杂原子的掺杂构型。杂原子掺杂石墨烯具有两个科学属性:其一,杂原子电负性异于碳原子时,掺杂会改变石墨烯的电荷分布,在其相邻碳原子位置形成结构缺陷和电荷的不均匀分布;其二,当杂原子电负性与碳原子相近而共价半径差异较大时,其掺入石墨烯会导致局部结构变化,从而增加电子的非对称自旋密度,形成有利于催化的活性位点95,96。以石墨烯为碳基体时,掺杂方式主要为原位掺杂和后处理掺杂,前者需要在制备石墨烯的同时掺杂杂原子,实现杂原子在碳骨架中的原位掺杂;后者则先制备出石墨烯前驱体,再利用含有杂原子的前驱体进行掺杂(如高温碳化、水热等)。
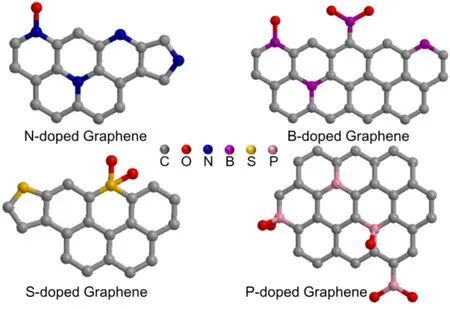
图5 杂原子掺杂构型示意图Fig.5 Illustration of doped graphene with different heteroatoms.
Ajayan等用化学气相沉积法,以甲烷为原料,Ni泡沫为模板合成三维石墨烯泡沫,并以g-C3N4为N源在高温下处理形成N掺杂石墨烯泡沫(NG)52。电子显微镜和拉曼光谱分析发现800 °C下处理获得的N掺杂石墨烯泡沫(NG-800)可保持碳的sp2杂化类型及良好的晶体结构。N掺杂构型可划分为吡啶N、吡咯N及石墨N三种,其中以吡啶N含量最多。将NG-800用于ECR,在-0.58 Vvs.RHE的电解电位下,CO的法拉第效率达到85% (图6a)。此外,他们还制备出N掺杂石墨烯量子点(NGQDs)并催化CO2还原为多碳产物53。具体策略是以氧化石墨烯(GO)为前驱体、二甲基甲酰胺为溶剂,利用溶剂热反应制备NGQDs。不同于催化产物为CO的一般N掺杂碳材料,NGQDs可将CO2还原为C2+产物。以1 mol·L-1KOH为电解液时,CO2还原的法拉第效率从18% (-0.26 Vvs.RHE)增加到90% (-0.75 Vvs.RHE),之后下降至64% (-1.03 Vvs.RHE)。在-0.26 Vvs.RHE电位下,只能检测到CO和HCOO-的产生,随还原电位不断增大,烃类(CH4、C2H4)以及多碳化合物(C2H5OH、CH3COO-、n-C3H7OH)生成。相比于无掺杂的石墨烯量子点,NGQDs还原电位减小的同时,对于C2+产物的选择性明显提高(图6b)。邹小龙与Ajayan等通过理论计算揭示了NGQDs催化CO2生成烃及含氧化合物的原因:当N原子掺杂进石墨烯量子点边缘位置时可以有效增强对于中间体*COOH的结合能,从而增强CO2向CO转化的能力。同时,吸附的*CH2与CO耦合形成C2产物(C2H4、C2H5OH)97。
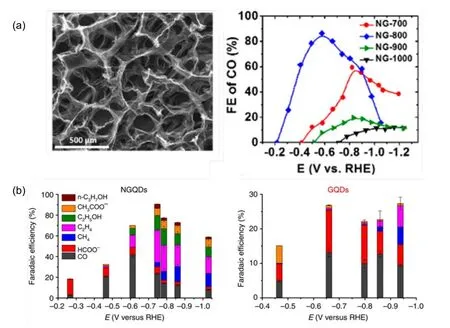
图6 (a)石墨烯泡沫SEM图及N掺杂石墨烯泡沫ECR性能52;(b)石墨烯量子点和N掺杂石墨烯量子点ECR性能53Fig.6 (a)An SEM image of graphene foam and Electrocatalytic activity of NGs towards CO2 reduction52,(b)Electrocatalytic activity of NGQDs and GQDs towards CO2 reduction53.
谭天伟等通过热解GO和三聚氰胺混合物制备N掺杂石墨烯(N-graphenes)63。N-graphenes对于甲酸盐的选择性达73% (-0.84 Vvs.RHE),虽然其甲酸盐选择性与一些金属催化剂(如nano-SnO2、In等98,99)相比尚存在差距,但其过电位大幅降低,降低了能耗。该催化剂吡啶N含量较高,其相邻的碳原子具有较多正电荷,因而展现出优异的催化活性。Phani等研究了B掺杂石墨烯(BG)在ECR中的应用100,用高温处理GO与硼酸的混合物,制备出B掺杂量约为4.1% (atomic fraction)的BG。相比于未经掺杂的石墨烯,BG活性提高了五倍以上。DFT计算表明B的掺杂增加了电子的非对称自旋密度,增强了对于CO2的吸附能力,促进了CO2的还原。
2.3 石墨烯基单原子催化剂
自张涛院士等在2011年首次制备出单原子催化剂——Pt1/FeOx,并将其用于CO氧化中以来,单原子催化剂以其高原子利用率、高活性和高稳定性等优点成为催化领域的研究热点101,102。碳基单原子催化剂是不同金属元素(M)以单原子的形式嵌入碳基体中,一般通过与N原子相互作用,在碳材料中形成M-N-C结构,因此,碳基单原子催化剂也可以称为金属掺杂碳材料。GO上存在大量且均匀分布的含氧官能团(―COOH、―OH等),这些含氧官能团由于静电相互作用而对金属阳离子具有较强的吸附能力,可以使金属阳离子均匀分布于GO表面,因此GO是一种比较理想的单原子催化剂基体材料。此外,相较于金属单质,金属单原子催化剂可以最大限度的提高其原子利用率;同时金属原子与碳基体之间存在较强的相互作用,因此具有良好的稳定性102,103。
常见的过渡金属钴(Co)、镍(Ni)等金属催化剂在HER中表现出较高活性;但当其以单原子形式存在时,CO2电还原活性则进一步提升,其催化产物一般为CO。2016年,Kamiya等热处理石墨烯与Ni的复合材料,得到具有Ni-N-C结构的催化剂,能够催化CO2还原为CO,法拉第效率最高超过90%64。汪淏田等以具有丰富缺陷的层状GO纳米片为碳基体吸附Ni离子,并用热处理得到Ni单原子催化剂65。层状GO纳米片表面有丰富的负电荷,有助于带正电荷的Ni在碳基体上均匀分布,同时这种2D结构使得活性中心密度达到最大,最终表现出较高的催化活性,对于CO的选择性最高达到95%。吴长征等用简易的离子吸附策略,制备出具有Ni-N4结构的高活性Ni催化剂(Ni2+@NG),通过高角度环形暗场扫描透射电子显微镜(HAADF-STEM)分析得到Ni以单原子的形式均匀分散于N掺杂石墨烯上,如图7a。对比于无Ni原子存在的石墨烯催化剂,Ni2+@NG催化剂的ECR活性大幅度提高,CO选择性达到92%66。蒋三平等用微波剥离方法,得到具有大面积和丰富缺陷的GO,并以此为基体材料实现了单原子Ni的高负载(达到6.9% (mass fraction))。同时,催化剂比表面积高达2380 m2·g-1。大的比表面积及高Ni原子负载量为CO2还原提供了丰富的活性位点,CO的选择性达到92.1%67。
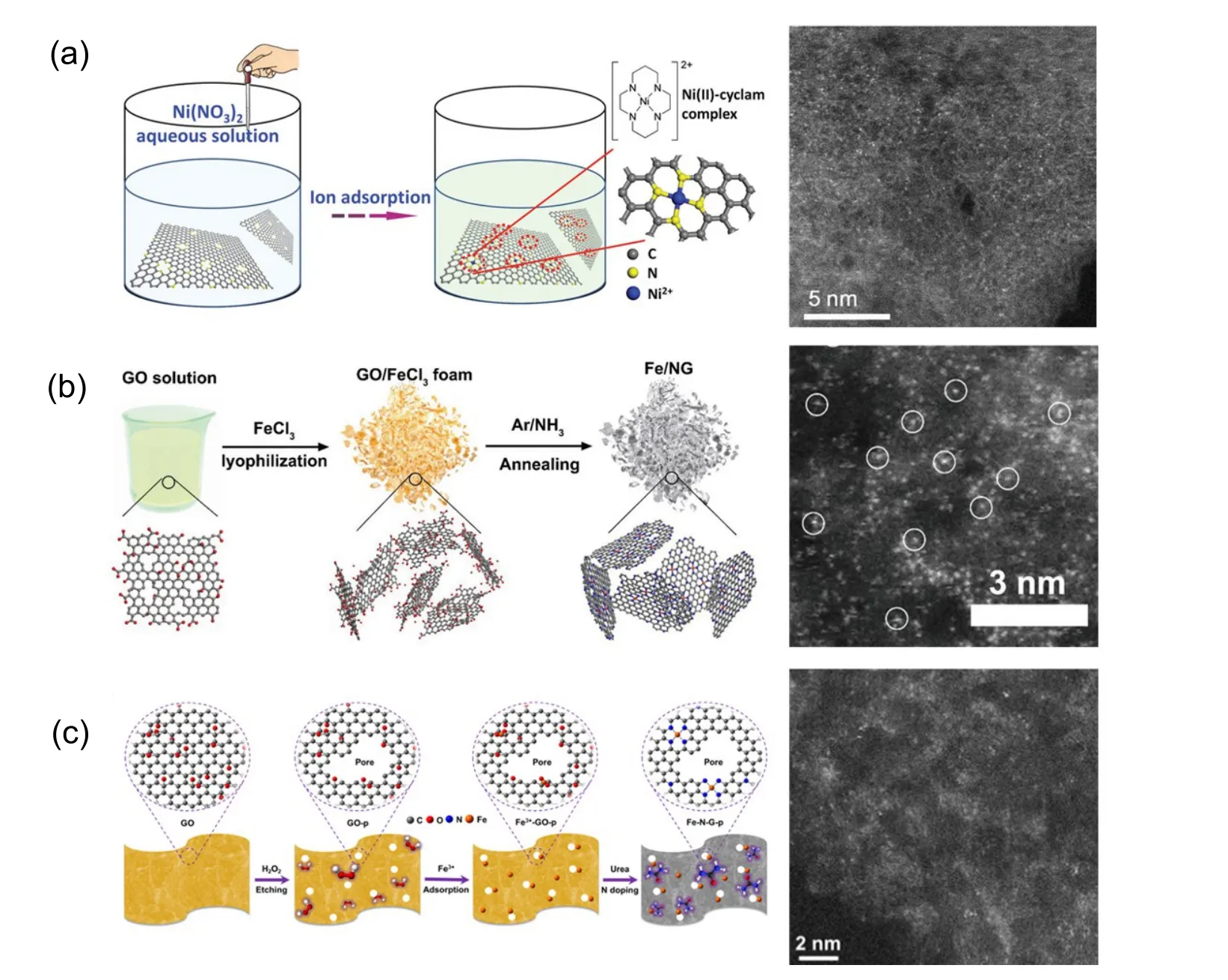
图7 (a)简易吸附方式制备Ni单原子催化剂66;(b)氮掺杂石墨烯锚定Fe单原子催化剂68;(c)石墨烯锚定N配位Fe单原子催化剂70Fig.7 (a)Surface immobilization of Ni Ions on N-doped graphene66,(b)Anchored Fe single atoms on N-doped graphene68,(c)Hosting atomically dispersed N-coordinated Fe sites on graphene70.
同样对HER有较好催化性能的铁(Fe)以单原子的形式锚定在碳基体上时,也展现出较好的CO2还原性能。Ajayan等将GO分散液与Fe离子溶液混合后经过冻干、热解等步骤,合成出分散良好的Fe单原子催化剂(Fe/NG),如图7b68。该催化材料中的Fe原子与四个N原子形成了有利于CO2吸附与活化的Fe-N4位点,实现了CO2向CO的转化,最高法拉第效率达到80% (-0.6 Vvs.RHE)。同时,发现催化剂中除Fe单原子位点以外,还有离散的N掺杂结构存在。DFT计算发现,Fe-N4结构周围带有两个石墨N结构时,反应势垒有更大幅度的下降,说明N的掺杂有利于提高催化剂的催化性能。王俊中等通过对氯化血红素和三聚氰胺分子在石墨烯表面的自组装和碳化处理,获得N掺杂石墨烯锚定的单原子Fe催化剂,热解过程中形成的锚定于碳基体中的FeN4位点会与N掺杂石墨烯中的吡咯N相互作用,形成了独特的FeN5构型。石墨烯的引入不仅增加了催化剂的导电性,同时比表面积也进一步增大,表现出较高的CO选择性(在-0.46 Vvs.RHE电位下,CO法拉第效率达到97%)及较低的过电位(350 mV)69。DFT计算表明吡咯N配位可有效抵消Fe 3d轨道的电子密度及降低Fe-CO的π反馈效应,有利于CO的快速解吸和高的选择性。德克萨斯A&M大学Li等用H2O2刻蚀GO,GO表面产生了能够吸附Fe3+的丰富纳米孔;将吸附有Fe3+的GO与尿素混合,经高温处理,得到Fe掺杂于石墨烯表面纳米孔位置的单原子催化剂,如图7c70。与未经H2O2刻蚀GO制备的Fe单原子催化剂比较,经H2O2刻蚀GO后制备的Fe单原子弱化了对于反应中间体*CO的吸附,更有利于CO的产生,其选择性最高可达到94%。
除Ni,Fe单原子催化剂对ECR表现出优异的电催化响应外,Sn、Zn、Bi、Cu、Mo、Mn、In等单原子催化剂相继被开发并呈现较高的催化活性17,71-75,104。谢毅院士等提出了一种实现Sn单原子催化剂公斤级制备的策略71;他们利用液氮急冷的方式,使Sn离子均匀分散于GO表面,经冷冻干燥得到固体材料,后续在氨气与氩气的混合气中热处理该材料制得Sn单原子催化剂。同步辐射表征发现,石墨烯表面原子分散的Sn带正电荷,有利于CO2的活化与质子化。与Ni、Fe单原子催化剂不同的是,Sn单原子催化剂对甲酸盐有较高选择性,法拉第效率可达74.3% (-1.6 Vvs.Saturated Calomel Electrode (SCE))。同时,键长相对较短的Sn―N键使得催化剂能够保持较高的化学稳定性,在200 h的连续电解中,ECR法拉第效率及电流密度基本没有下降。2019年,谢毅等制备出一种钼(Mo)单原子催化剂,以离子液体为电解液时,同样可以实现将CO2还原为甲酸盐,其选择性相比于无Mo原子存在的N掺杂石墨烯提高了一倍104。
3 石墨烯复合催化剂
在石墨烯的基面修饰活性组分(如过渡金属化合物、碳量子点等),是提升石墨烯整体电催化活性的另一关键策略。通过与石墨烯复合,存在缺陷的活性组分(如稳定性较差、形貌及结构不能很好地保持的金属单质及金属化合物,导电性相对较低的碳点等非金属材料)可以在一定程度上克服这些缺陷,有效增强导电性,防止活性组分团聚,从而提高整体催化性能及稳定性。以ECR的金属催化剂Cu2O为例,虽具有较高乙烯选择性,但其稳定性往往较差,易于团聚而失活,这些问题在一定程度上将阻碍ECR的规模化应用105。因此耦合石墨烯的高导电性与金属催化剂的高活性,采用多元构筑策略(包括碳包覆制备核壳结构、石墨烯表面锚定等)创制石墨烯-金属复合材料,利于提升ECR性能。
3.1 石墨烯-金属单质复合催化剂
超细Au纳米颗粒(约2.4 nm)凭借其高质量活性在ECR中展现出了应用潜力,但其丰富的低配位位点有利于HER从而降低对ECR的产物选择性76,106,107。澳大利亚伍伦贡大学Wang等以还原氧化石墨烯(rGO)片为载体,用胺物种对Au纳米颗粒表面进行修饰,胺物种在提高对CO2的吸附能力的同时可以有效抑制HER,很好的解决了Au催化ECR的低产物选择性问题,另外胺分子修饰的Au纳米颗粒仍保持了超高的质量活性(> 100 A·g-1)76。通过采用不同胺分子修饰及用乙醇去除氨基,并比较去除前后复合催化剂的电化学性能,发现线性胺的修饰有利于CO2转化为CO,但由于氨基与Au位点之间的强相互作用及胺分子构型的影响,增长烷基链能够降低Au位点上的氨基覆盖率从而更有利于ECR进行;而支链胺由于具有更高的氨基基团密度不利于CO的产生。Fischer等基于自下而上策略制备了一系列的三维石墨烯纳米带网络(GNRs),其结构因小分子前体不同而有所差异(图8a,b)77。GNRs基底与金属之间的强相互作用能够防止Au纳米颗粒(Au NPs)的聚集,提高了嵌入网络中的Au NPs的分散性,从而有效改善催化剂的长期稳定性并提高其催化反应活性。他们研究了GNRs对Au NPs催化性能的影响,发现具有较低功函数的cove-type GNRs能够促进电子由载体界面向Au NPs的转移,增加金属表面的电荷密度,进而降低ECR起始电位。朱俊杰等以十六烷基三甲基溴化铵(CTAB)为表面活性剂,用晶体生长法,制备出尺寸约60 nm的单晶Au纳米颗粒(SCAu),再经水热将Au表面的CTAB转化为N掺杂石墨烯量子点(NGQDs),暴露出更多的Au表面,提高了催化剂的整体活性78。Bader电荷分析结果表明SCAu与NGQD之间存在由SCAu向N掺杂石墨烯的电子转移,导致电子在N掺杂石墨烯上富集,从而有效降低吡啶N位点上由CO2还原为CO的速控步骤(形成*COOH中间体)的能垒,提高了NGQDs-SCAu NPs复合催化剂的整体活性,在较低的电位下CO法拉第效率达93%。
图8 (a)小分子前体合成chevron type GNRs;(b)小分子前体合成cove type GNRs77Fig.8 (a)Synthesis of chevron GNRs from molecular precursors,(b)Synthesis of cove GNRs from molecular precursors77.
虽然Au、Ag、Pd、Ga等金属对ECR有非常高的本征活性,但此类材料在自然界中储量稀少、成本昂贵。而金属Zn储量相对丰富,其衍生物、复合物成本较低,可作为上述贵金属催化剂的替代材料108-110。Hwang等将rGO层作为传质屏障包裹在多孔锌电极外提高了CO选择性33。由于HER与ECR受传质影响效果不同,随着rGO在锌电极表面的覆盖率提高,材料表界面处质子的快速消耗使得电解液中质子浓度过低,导致电解过程中pH增加的程度高于pCO2,故在锌电极表面引入rGO在抑制HER的同时提高了CO的法拉第效率(图9)。原位X射线吸收光谱分析发现rGO层下Zn的化学态没有明显改变,说明rGO层对金属态Zn有保护作用。相似地,将CO2还原为甲酸盐的金属Bi、Sn等无毒且价廉金属也是Cd、Pb等重金属的代替材料。鄢俊敏等以硼氢化钠为还原剂,首次利用还原的方式,将超细Bi纳米颗粒锚定于rGO上(Bi/rGO)79。Bi与rGO之间存在电子相互作用,有利于对CO2和CO2·-的吸附从而促进ECR过程。同时,与利用聚乙烯吡咯烷酮为分散剂制备的具有相似粒径的Bi纳米颗粒对比,由于Bi/rGO没有分散剂对活性位点的掩蔽而具有更大的电化学活性表面积,因而暴露出更多的活性位点。这种复合物催化剂对甲酸(HCOOH)展现出高的选择性,在较低的电位下(-0.8 Vvs.RHE)HCOOH法拉第效率能达到98%。

图9 计算的局部pH及还原氧化石墨烯的覆盖效应:(a)计算流体动力学模拟示意图;(b)CO,H2法拉第效率与石墨烯覆盖率关系;稳定状态下,石墨烯覆盖率分别为0、50%、97.5%的pH (c)和pCO2 (d)33Fig.9 Computed local pH and reduced GO coverage effects.(a)Schematic of computational fluid dynamics simulation domain,(b)Plots of CO FE and H2 FE as a function of coverage level,Contour plots of (c)pH and(d)pCO2 under a steady state at different coverage levels of 0,50%,and 97.5%,respectively33.
Cu可将CO2还原为多碳产物,通过构建不同形貌、不同组成或与其他金属、非金属复合构筑Cu基催化剂已经实现将CO2转化为CO、CH4、CH3OH、C2H5OH以及C2H4等111-114。孙守恒等将富吡啶N掺杂石墨烯(p-NG)与Cu纳米颗粒复合,由于吡啶N呈路易斯碱性,所制备材料能够有效吸附并稳定CO2分子,促进附近Cu位点上的*COOH中间体的质子化及进一步的C―C耦合,进而提高了对多碳产物的选择性80。在-0.9 V (vs.RHE)电位下,C2H4的法拉第效率为19%,占总碳氢产物的79%。韩布兴院士等将N掺杂石墨烯量子点(NGQ)与CuO衍生的Cu纳米棒(Cu-nr)复合,由于NGQ具有亲氧性,有利于对含氧中间体如*CH2CHO的吸附和稳定,促进了*CH2CHO进一步质子化转化为C2H5OH81。DFT计算也表明NG/Cu(111)表面明显降低了*CH2CHO质子化形成*C2H4O的能垒,比*CH2CHO转化为乙烯路径更有利,热力学上更倾向于产醇。拉曼光谱分析显示复合催化剂和纯Cunr表面的Cu―CO、C≡O信号峰强度接近,且两者均能产生多碳产物,故NGQ和Cu-nr能提供双活性位点,两者之间的协同作用促进ECR沿着产醇的路径进行。最终,该催化剂对于C2及C2+的醇类的选择性达到52.4% (-0.9 Vvs.RHE),远高于未复合NGQ的Cu-nr。此外,该催化剂也表现出极佳的稳定性,连续电解100 h,产物法拉第效率无明显变化。
3.2 石墨烯-金属化合物复合催化剂
目前,ECR的金属基催化剂不仅仅集中于单质金属,金属的化合物形态往往也表现出良好的催化活性。王立世等以正丁胺为氮源、乙酰丙酮钴为金属前驱体,用水热法制备出负载部分被氧化的Co纳米颗粒的富含吡咯N的单层石墨烯(PO-5 nm Co/SL-NG)82,该复合催化剂还原CO2产生甲醇的优异性能源于载体石墨烯与金属之间的协同作用,即PO-5 nm Co具有催化CO2还原为甲酸盐、甲醇的活性,而SL-NG能促进多质子转移加氢过程,且其表面存在大量的褶皱结构,这有利于金属纳米颗粒的均匀分散。该复合物有利于多质子耦合电子转移,提高甲醇的选择性。鉴于GO中的碳原子存在大量孤对电子,在π-π共轭作用的影响下GO表面带负电荷,严乙铭等利用这一特性,将带正电荷的Cu(II)吸附在GO表面,经热处理和H2还原并调控还原时间,制备了CuxO修饰的GO复合材料(G-CuxO-T)83;其中,经过2 h处理得到的GCuxO-2h还原产生HCOOH的性能最佳,在-0.8 V(vs.RHE)电位下的选择性达到81%,9 h内稳定性良好;G-CuxO-2h对产物HCOOH的高选择性可能源于其表面更高的Cu(I)含量,Cu(I)有利于CO2·-中间体的稳定、促进速控步骤——HCOO-解吸过程的发生。经过长时间工作之后其活性多有衰退;这是由于催化剂活性组分的重构、聚集以及催化剂的价态、组成和形貌等性质改变造成的。G-CuxO-2h显示出良好稳定性的原因是其中存在一种“缓冲效应”:电解CO2过程中,G-CuxO-2h中的Cu(I)在被逐渐还原为Cu(0)的同时,其表层中的Cu(II)可接收电子转化为Cu(I),从而维持了催化剂表面固定含量的Cu(I)活性位点,进而赋予催化剂的良好活性与稳定性。提高催化剂稳定性的一个重要方式就是维持活性位点的数目。王海梁等提出的一种自清洁催化剂电极表现出良好的稳定性。他们将Pd原子预先沉积在催化剂表面,在电解过程中,Pd原子诱导催化剂表面形貌与组成的连续重组,从而不断更新催化剂表面,最终表现出良好的稳定性20。Officer等将具有烷氧基取代的Co酞菁(CoPc-A)与化学转化石墨烯(CCG)复合,具有位阻效应的烷氧基取代的Co酞菁有效避免了Co酞菁在石墨烯表面的聚集,维持了单个分子的催化活性,从而拥有优异的稳定性89。
Amal等以碳纤维纸为基体,用等离子增强化学气相沉积的方式,制备了3D垂直缺陷石墨烯(VG-Ar),再采用电沉积的方式将Cu纳米颗粒沉积到其表面,制得Cu/VG-Ar复合材料84。X射线光电子能谱(XPS)及Cu俄歇谱分析证实,VG-Ar表面的铜物种以Cu、Cu2O和CuO形式存在且以Cu2O为主,VG-Ar对Cu2O的稳定起重要作用;原位同步粉末衍射等表征及DFT计算也表明VG-Ar中的sp3本征缺陷能够稳定Cu2O物种。同时石墨烯的三维垂直结构为Cu活性物种提供了丰富的锚定位点,从而增强对CO2分子及其中间体的吸附与稳定,抑制HER活性的同时提高了碳氢产物的选择性。曾杰等将GO与In前体通过水热法处理,制备出In(OH)3-rGO异质结构,再在空气中退火处理,得到In2O3与rGO的复合材料85。rGO与多孔In2O3纳米带之间的化学耦合作用导致电子由rGO向In2O3转移,一方面降低了材料的界面电荷转移电阻,另一方面也降低了关键中间体*HCOO-形成所需的吉布斯自由能,从而大幅度提高其催化CO2还原为甲酸盐的性能。刘立成等将0D掺N石墨烯量子点(NGQDs)与2D氧化铋(Bi2O3)纳米片复合,在-0.9 --1.2 Vvs.RHE的宽电势范围内,对甲酸盐的法拉第效率平均约为95.6%。DFT计算结果表明,这种宽电势范围内的高活性是因为NGQDs极大地增强了材料对CO2(ads)和*OCHO中间体的吸附能力86。
以过渡金属复合物为代表的分子催化剂,结构多样且含有均匀、明确、可调的活性位点,具有较高的选择性。把分子催化剂固定在石墨烯基导电基底上,能够使两者优势相结合从而表现出更加优异的性能。王昕等将平面Co复合物(CoII-2,3-naphthalocyanine complexes)锚定在不同元素掺杂的石墨烯基底上,得到多相Co电催化剂,能够选择性地将CO2还原为CO87。通过调节前驱体,制备了N、O、S等不同元素及官能团掺杂改性的石墨烯基底,发现石墨烯的亚砜(SO)掺杂和羧基(COO)掺杂结构能够通过轴向配位锚定Co复合物,进而作为CO2电还原的主要催化活性位点。与羧基相比,亚砜基能进一步促进Co位点与石墨烯基底之间的电子传输,从而提高催化活性。将1,10-phenanthroline与Cu2+配位而成的Cu复合物锚定到介孔石墨烯基底上,处于扭曲的三角双锥结构中心的五配位Cu(I)是主要的催化活性物种,在外加电场作用下Cu复合物能够可逆地发生异质化,致使其电子密度增加,而石墨烯的高表面孔隙率限制了由体相溶液到电极表面的质量传输,从而有效地抑制了HER88。Officer等将具有烷氧基取代的Co酞菁(CoPc-A)与化学转化石墨烯(CCG)复合(CCG/CoPc-A)以实现CO2向CO催化转化89。CoPc-A分子通过π-π相互作用固定到石墨烯上,拉曼光谱分析发现CoPc-A与CCG的复合几乎没有改变CCG的ID/IG值,而CCG/CoPc的ID/IG明显增大(图10a),可能是未经烷氧基取代的Co酞菁与石墨烯之间存在较强的相互作用,从而显著影响了CCG的芳香度,改变了其ID/IG。而具有位阻效应的烷氧基的取代能够有效缓解Co酞菁在石墨烯上的堆积,显著增强了单个酞菁分子的催化活性(图10b)。与CCG/CoPc相比,CCG/CoPc-A复合催化剂在480 mV的过电位下进行连续30 h的电解测试时,其电流密度以及CO的法拉第效率无明显下降,表现出良好的稳定性。

图10 (a)CCG、CoPc-A、CCG/CoPc-A及CoPc、CCG/CoPc的拉曼光谱图;(b)CCG/CoPc和CCG/CoPc-A的CO2催化机理图89Fig.10 (a)Raman spectra of CCG,CoPc-A,CCG/CoPc-A hybrid,CCG,CoPc,and the CCG/CoPc hybrid,(b)Proposed CO2 catalysis mechanism on the CCG/CoPc and CCG/CoPc-A hybrids89.
4 总结与展望
利用电化学技术,将CO2转化为液态燃料或高附加值的化学产品,有广阔的发展前景。这可以实现有害温室气体CO2的资源化利用,减少对人类社会与环境的危害,实现人造碳循环。碳基催化剂在CO2的还原中有潜在的重要应用前景,与贵金属催化剂相比,成本更加低廉,对特定产物的选择性与催化稳定性更好。石墨烯作为碳材料的重要组成部分,通过制造表面缺陷或表面改性等方式,可调变石墨烯的电子、原子结构,提升其电催化CO2还原活性。以石墨烯为催化剂载体,不仅可以增加催化剂的导电性,还可以在保留其本身优势的同时与负载的金属基催化剂协同提高催化剂活性。
迄今,石墨烯基电催化剂已取得较大进展,但仍存在一些挑战性问题:1)电流密度较低,过电位较大,现阶段ECR过程能达到的电流密度不超过100 mA·cm-2(使用传统的H型电解池时),即使对电解系统优化(使用气体扩散电极、流通池等)之后也难以实现大于500 mA·cm-2的工业级电流密度,且较大的过电位使得ECR过程能量效率较低;2)在石墨烯改性过程中,引入单一掺杂构型、制备特定缺陷结构催化剂较为困难;3)目前制备的催化剂多为克级甚至毫克级,需要实现千克级规模化的制备;4)稳定性则是ECR未来规模化应用需要考虑的重点因素之一。目前,ECR催化剂稳定性多在10-30 h,而工业化发展则需要催化剂稳定性达到1000 h以上;5)反应机理尚不清楚或存在一定的争议,表现为同一种类型的催化材料有不同的反应机理,如对N掺杂碳材料来说,吡啶N、吡咯N以及石墨N均被不同工作证实对ECR有利。
高性能石墨烯基ECR催化剂的设计与构筑,是一个富有挑战性的工作,需要从以下几个方面着手:1)改性石墨烯。原始石墨烯中的碳原子呈电中性,ECR活性低,很难活化CO2分子及吸附反应中间体;通过掺杂杂原子、制造空位缺陷及修饰官能团等,可以提高石墨烯的活性。需要探究石墨烯的高效改性策略;2)控制形貌。催化剂的形貌对于ECR的性能影响很大,不论是石墨烯直接用作催化剂活性组分还是作为载体,不同维度的石墨烯材料(0D石墨烯量子点、1D石墨烯纳米带、2D石墨烯及3D石墨烯泡沫等)和不同孔结构(微孔、介孔、大孔及分级孔结构)对催化剂活性位点的暴露、电子转移及CO2的传质等均有显著影响;3)引入金属活性组分。石墨烯作为理想的载体材料,与其他活性组分的复合是进一步提升石墨烯ECR活性的有效途径。此外,石墨烯与金属组分之间存在的强金属-载体相互作用可避免金属活性组分的不可逆聚集,进而提高催化剂稳定性。4)制备石墨烯基整体材料。传统意义上制备出的石墨烯等碳基催化剂多为粉体材料,因此在制作电极时必须用到一些有机粘结剂(如5% Nafion溶液),将催化剂粉末负载于导电基体上;粘结剂的使用会减弱催化活性物质和电极的接触,在一定程度上影响催化活性和催化剂的稳定性。利用石墨烯易于制备成凝胶材料或泡沫石墨烯等特点制成尺寸合适的电极材料,降低粘结剂的不利影响;5)反应机理的诠释。弄清CO2还原反应机理对于反应活性位点的识别、反应催化剂的设计及反应产物的控制等至关重要。需要有机融合实验和理论计算,综合采用in situ和operando表征技术,深入理解反应机理,有助于设计出新结构高效的碳基电催化剂。
