First-principles study of two new boron nitride structures:C12-BN and O16-BN
2022-02-24HaoWang王皓YaruYin殷亚茹XiongYang杨雄YanruiGuo郭艳蕊YingZhang张颖HuiyuYan严慧羽YingWang王莹andPingHuai怀平
Hao Wang(王皓) Yaru Yin(殷亚茹) Xiong Yang(杨雄) Yanrui Guo(郭艳蕊)Ying Zhang(张颖) Huiyu Yan(严慧羽) Ying Wang(王莹) and Ping Huai(怀平)
1College of Science,Civil Aviation University of China,Tianjin 300300,China2Center for Transformative Science,ShanghaiTech University,Shanghai 201210,China3Institute of Applied Physics,Chinese Academy of Sciences,Shanghai 201800,China4Shanghai Synchrotron Radiation Facility,Shanghai Advanced Research Institute,Chinese Academy of Sciences,Shanghai 201210,China
Based on the first-principles method,we predict two new stable BN allotropes: C12-BN and O16-BN,which belong to cubic and orthorhombic crystal systems,respectively. It is confirmed that both the phases are thermally and dynamically stable. The results of molecular dynamics simulations suggest that both the BN phases are highly stable even at high temperatures of 1000 K. In the case of mechanical properties, C12-BN has a bulk modulus of 359 GPa and a hardness of 43.4 GPa, making it a novel superhard material with potential technological and industrial applications. Electronic band calculations reveal that both C12-BN and O16-BN are insulators with direct band gaps of 3.02 eV and 3.54 eV,respectively. The XRD spectra of C12-BN and O16-BN are also simulated to provide more information for possible experimental observation. Our findings enrich the BN allotrope family and are expected to stimulate further experimental interest.
Keywords: new boron nitride phase,first-principles calculations,mechanical properties,electric properties
1. Introduction
As the chemical analogue of carbon, novel boron nitride (BN) allotropes have attracted extensive theoretical and experimental attentions for their fascinating properties. BN shares similar structures with carbon from zero-dimensional(0D) buckyball,[1,2]one-dimensional (1D) nanotubes and nanoribbons,[3,4]two-dimensional (2D) nanosheets,[5]to three-dimensional (3D) crystalline or amorphous BN.[6]For the 3D crystalline structures,BN allotropes can be mainly divided into two groups. One group includes diamond-like c-BN and w-BN constituting the sp3bonding network,[7]while the other one is graphite-like h-BN and r-BN containing only sp2bonds. It is generally recognized that c-BN is a thermodynamically stable phase under ambient conditions and h-BN becomes stable at much higher temperature over 1200 K. In spite of the structural similarity between BN and carbon,their physical and chemical properties are thoroughly different because of the ionic character in B—N bonding.Due to the difference in electronegativity,N and B atoms form strongly polarized covalent bonds,leading their BN compounds to fascinating mechanical[8]and thermal[9]properties and excellent tolerance under extremely harsh environment such as high temperature and high pressures.
The similarity in structure with diverse properties compared to carbon materials has attracted intensive experimental and theoretical attentions to explore more novel BN structures.Recently, based on the concept that all the carbon allotropes predicted previously may have one-to-one counterparts in BN allotropes, a lot of novel BN structures have been proposed and studied, such as bct-BN, Z-BN, M-BN, P-BN, H18-BN,Rh6-BN,O-BN,T-B3N3,and HCBN,[10—16]corresponding to Bct-C4,Z-carbon,M-carbon,P-carbon,H18carbon,Rh6 carbon,oC32 carbon,T6 carbon,and CHC,[17—25]respectively.In addition,there are some other novel carbon and BN structures,such astP40carbon, C48 carbon and B4N4.[26—28]Among those BN allotropes, Z-BN and O-BN are confirmed to be novel superhard materials owing to the high bulk modulus and Vickers hardness, based on the first-principles calculations.One of these materials, i.e., T-B3N3, proposed by Zhanget al., is the first reported 3D BN structure with intrinsic metallicity. It is a tetragonal phase consisting of a mixed sp2and sp3bonding network. Another BN structure, i.e., HCBN, a hexagonal phase containing only sp2bonds, is also predicted to exhibit intriguingly intrinsic metallicity in succession. In addition, Xionget al.proposed an sp2and sp3-hybridized metallic monoclinic 3D BN structure, which is confirmed to be the most energetically favorable structure among the previously predicted metallic BN structures.[29]However, to the best of our knowledge,most of the BN allotropes and their carbon counterparts have not been synthesized experimentally.
Recent shock compression experiments of polycrystalline graphite produced obvious evidence of a new carbon phase.Liet al.have theoretically confirmed it to be a body-centeredcubic carbon phase termed BC12.[30]Wanget al.identified a novel topological semimetal carbon phase termed bco-C16in all sp2bonding networks byab initiocalculations.[31]The simulated x-ray diffraction spectrum matches well with the measured XRD spectra of detonation and chimney soot, indicating the presence of bco-C16in the specimen. Under the inspiration of BC12 and bco-C16carbon,we propose two new BN allotropes termed C12-BN and O16-BN, which are analogous to the recently reported BC12 and bco-C16carbon, respectively. In this paper, first-principles calculations are conducted to illustrate the physical properties of C12-BN and O16-BN.Firstly,the energetic and dynamical stability are confirmed with respect to the volume-dependence of total energy per atom, cohesive energy and phonon dispersion. Secondly,the mechanical properties are studied based on the calculations of the elastic constants. The Vickers hardness of C12-BN is obtained to be 43.4 GPa, indicating that it is a superhard material. The mechanical properties of O16-BN are relatively poor. However,O16-BN has low density and porous structure,which leads to potential applications in adsorption materials,e.g., hydrogen storage. In addition, the analysis of electronic properties reveals that both C12-BN and O16-BN are insulators with direct band gaps of 3.02 and 3.54 eV, respectively.Their XRD spectra are also provided for further experimental identification.
2. Computational methods
The structural optimization and properties of two new BN allotropes are calculated using the density functional theory(DFT)as implemented in the Viennaab initiosimulation package (VASP),[32]with projector augmented wave (PAW)potentials.[33]The exchange—correlation of electrons is treated within the generalized gradient approximation (GGA) in the form of Perdew—Burke—Enzelhof (PBE) functional.[34]Plane waves with kinetic energy cutoff of 600 eV are adopted to expand the wave function of valence electrons(2s22p1for B and 2s22p3for N). The crystal structures are optimized using the conjugate gradient method, in which the convergence criteria for the electronic and the ionic relaxation are set to 10−7eV and 10−5eV/for energy and force, respectively. These choices ensure that all the enthalpy calculations are well converged to less than 1 meV/atom. The Brillouin zone sample meshes for all mentioned systems are set to be dense enough(9×9×9 for c-BN, 19×19×9 for h-BN, 11×11×11 for BC8-BN,9×9×9 for C12-BN,and 7×11×17 for O16-BN).The energetic and thermodynamic stabilities of these BN allotropes can be evaluated by comparing their total energies and cohesive energies. Based on these optimized structures, we calculate their elastic constants using the strain-stress method.The bulk modulus, shear modulus and Vickers hardness are then derived from the Voigt—Reuss—Hill averaging scheme.[35]The phonon frequencies for C12-BN and O16-BN are calculated using the direct supercell method,which uses the forces obtained by the Hellmann—Feynman theorem implemented in the supercell. Phonon spectra calculations are then carried out by diagonalizing the dynamical matrix using the Phonopy package.[36]Electronic band structures are calculated using the Heyd—Scuseria—Ernzerhof hybrid functional(HSE06).[37]
3. Results and discussion
As shown in Table 1,we first present the structural characters of the two new BN phases.The crystal structure of C12-BN belongs to theI¯43dspace group.
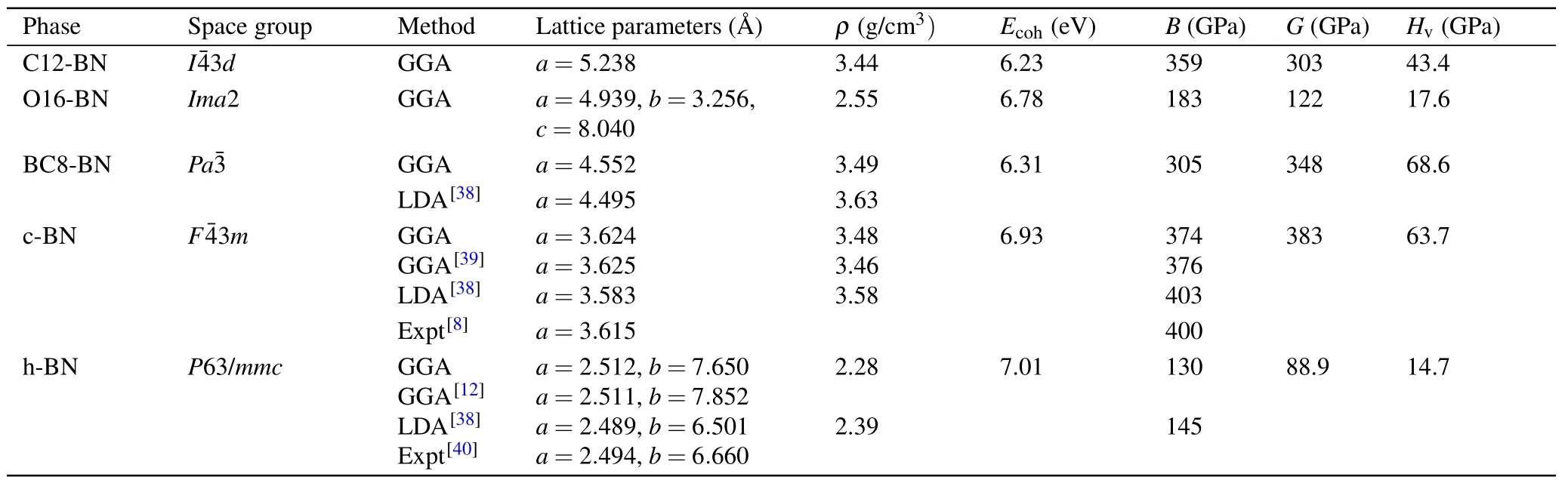
Table 1. The calculated lattice parameters a,b,and c,density(ρ),cohesive energies(Ecoh),bulk modulus(B),shear modulus(G)and Vickers hardness(Hv)for C12-BN,O16-BN,BC8-BN,c-BN and h-BN.
In cubic representation depicted in Fig. 1(a), its equilibrium lattice constant derived from GGA isa=5.238with N atoms occupying 12b(0.5, 0.25, 0.125) and B atoms occupying 12a(0.75, 0.125, 0) Wyckoff positions. Figure 1(b)shows the primitive unit cell of C12-BN, in which there are 12 atoms with lattice parameters optimized to bea=4.536,α=β=γ=109.47°. All the B—N bonds in C12-BN are sp3bonded and have equal length of 1.604, which is slightly larger than that of diamond-like c-BN (1.570). However,there are two kinds of bond angels, i.e., 131.81°and 99.59°in C12-BN, which are also found in a body centered cubic phase BC8-BN as 115.74°and 102.09°, respectively. The new orthorhombic phase O16-BN belongs to theIma2 space group. At zero pressure, its equilibrium lattice constants area= 4.939,b= 3.256andc= 8.040. There are 16 atoms in the orthorhombic unit cell of O16-BN,in which two nonequivalent N atoms occupy the 4a(0.0, 0.0,−0.4096)and 4b(0.25,−0.6259,−0.1757), and two nonequivalent B atoms occupy the 4a(0.0,0.0,−0.5882)and 4b(0.25,0.1256,−0.3265)Wyckoff positions. As shown in Fig.1(d),only sp2configuration exists in the O16-BN structure, which is different from the all sp3B—N bonding characters in C12-BN.There are four inequivalent bonds in O16-BN,with their bond lengths of 1.457, 1.463, 1.480, and 1.436, respectively. The average bond length is 1.459,which is comparable with the sp2bond length in h-BN(1.45).
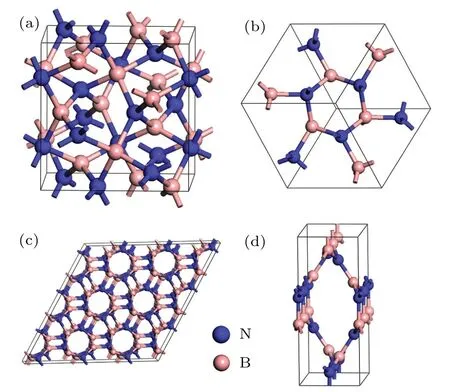
Fig.1.(a)The cubic crystal structure of C12-BN in I¯43d symmetry.(b)The primitive cell of C12-BN containing twelve atoms.(c)A hexagonal representation of C12-BN.(d)The orthorhombic unit cell of O16-BN.
To understand the energetic stabilities of C12-BN and O16-BN, we calculated their total energies as a function of volume using the GGA scheme. Figure 2 shows the calculated total energy per atom versus its volume for C12-BN and O16-BN, together with those of other reported BN allotropes. It is interesting to see that theE—Vcurve of C12-BN is very close to that of BC8-BN,and the equilibrium volume per atom is comparable with that of sp3bonded c-BN and BC8-BN.Further,we can see that C12-BN is more stable than M-BN.Therefore,the C12-BN phase is almost as stable as BC8-BN. However, it can be obviously found that O16-BN is less stable than the most three stable phases of c-BN,h-BN and Z-BN, but more stable than other BN structures.The equilibrium volume is larger than that of c-BN and closer to that of h-BN, which is reasonable due to the sp2bonding characters in O16-BN. To further investigate the thermodynamic stabilities of C12-BN and O16-BN, we calculate their cohesive energies, which are the main parameters to measure thermodynamic stability. The cohesive energy is defined asEcoh=[E(BN)−nBE(B)−nNE(N)]/nTotal, whereE(BN) is the total energy of the unit cell of BN structure,n(B/N)is the number of B/N atoms in the unit cell, andE(B/N) is the energy of a single pure B/N atom. The results are summarized in Table 1,with those of c-BN,h-BN and BC8-BN also provided for comparison. The cohesive energy of C12-BN is obtained to be 6.23 eV,being only 0.08 eV slightly less than BC8-BN.For O16-BN,the cohesive energy is 0.47 eV larger than BC8-BN and 0.15 eV less than c-BN.Therefore,both the results of total energy and the cohesive energy indicate that C12-BN and O16-BN have similar stabilities as other stable BN allotropes reported before.
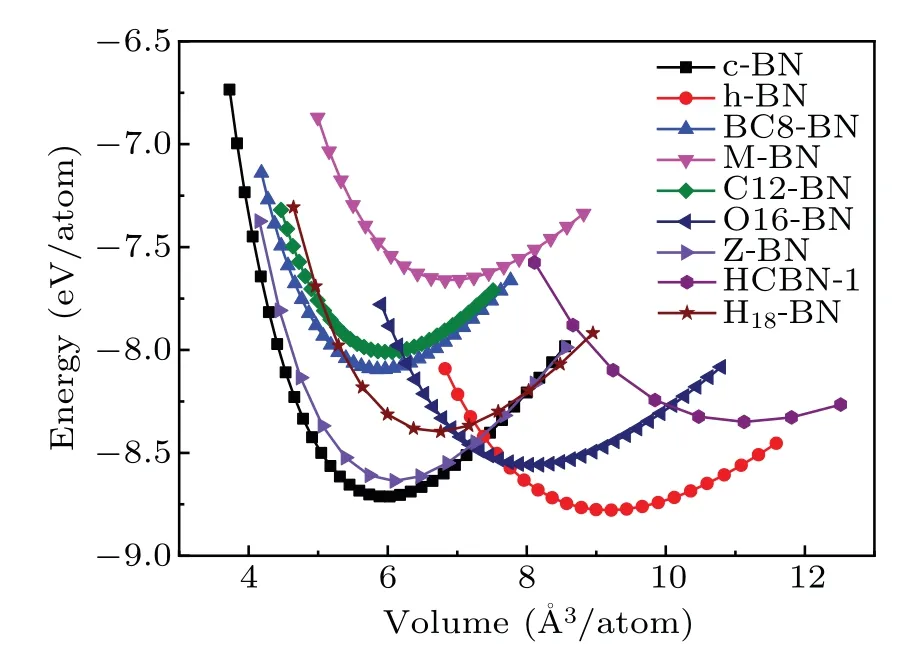
Fig.2. Total energies as a function of volume per atom for C12-BN and O16-BN in comparison with other reported BN structures.
The phonon dispersion plays an important role in examining the dynamical stability of a structure. The appearance of imaginary frequencies in the phonon dispersion spectra leads to unstable structures. Therefore,we examined the dynamical stability of C12-BN and O16-BN by calculating their phonon dispersion curves at zero pressure. As shown in Fig. 3, no imaginary frequencies were observed throughout the whole Brillouin zone, confirming the dynamical stabilities of both C12-BN and O16-BN.

Fig.3. Calculated phonon band structures of C12-BN(a)and O16-BN(b)at zero pressure.
In order to investigate the mechanical stabilities and properties of C12-BN and O16-BN,we calculate their elastic constantsCij. To ensure mechanical stability, all elastic constants should satisfy the well-known Born—Huang stabilitycriteria. As a cubic crystal, the three independent elastic constants (C11= 725.4 GPa,C12= 176.7 GPa andC44=322.8 GPa) of C12-BN satisfy the stability criteria (C11>0,C44>0,C11> |C12|andC11+2C12>0), suggesting that the C12-BN phase is mechanically stable. For orthorhombic phase structures, the mechanical stability criteria is given byC11>0,C22>0,C33>0,C44>0,C55>0,C66>0,C11+C22+C33+2(C12+C13+C23)>0,C11+C22−2C12>0,C11+C33−2C13>0,C22+C33−2C23>0. In our results,the calculated nine independent elastic constants for O16-BN are given asC11=624.7 GPa,C22=789.4 GPa,C33=107.0 GPa,C44=71.5 GPa,C55=73.1 GPa,C66=277.7 GPa,C12=231.6 GPa,C13= 131.2 GPa, andC23= 61.9 GPa. Obviously, they satisfy the mechanical stability criteria. Based on the elastic constants, we can also evaluate the bulk modulus(B),shear modulus(G)and Vickers hardness(Hv),whereHv=2(G3/B2)0.585−3. As for the C12-BN phase, it is obtained thatB=359 GPa,which is slightly lower than that of c-BN(376 GPa)and higher than that of BC8-BN and h-BN.The value of Vickers hardness is 43.4 GPa, indicating that C12-BN is a potential superhard material(Hv>40 GPa). Although the bulk modulus and Vickers hardness of O16-BN are lower than those BN allotropes consisting of sp3bonds,they are still higher than that of h-BN. Due to the low density and porous structure,O16-BN may have potential applications in adsorption materials.
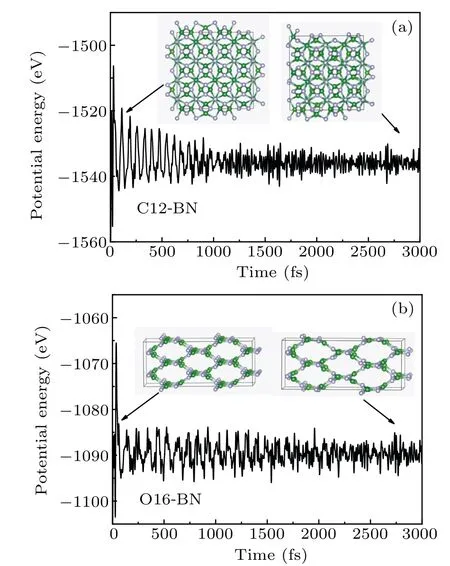
Fig.4. MD simulations on the potential energy fluctuations of(a)C12-BN and (b) O16-BN at 1000 K. Insets show the crystal structures at certain time steps.
The thermal stabilities of C12-BN and O16-BN are examined by means of MD(molecular dynamics)simulations with the NVT ensemble. The temperature is set as 1000 K. The simulated system is 2×2×2 supercell and the time step is chosen to be 1 fs. The MD simulations are carried out on the supercell of both C12-BN and O16-BN for 3000 time steps,which is long enough to reach steady state.As shown in Fig.4,the structures of C12-BN and O16-BN are still kept intact at 1000 K with 3000 time steps. There is also no drastic transformation in the potential energy fluctuation curves,indicating that no structure transformation happens. The results suggesting that both C12-BN and O16-BN are highly stable even at high temperatures of 1000 K.Therefore,the thermal stabilities of C12-BN and O16-BN are confirmed.

Fig. 5. Calculated electronic band structures and density of states(DOS) in arbitrary units for C12-BN (a) and O16-BN (b). The Fermi level is set to zero and denoted by the dashed line. The valence band maximum and conduction band minimum of C12-BN and O16-BN are both located at the Γ point with a direct band gap of 3.02 and 3.54 eV,respectively.
In addition, to explore the electronic properties of C12-BN and O16-BN,we calculate their electronic band structures and densities of states. As shown in Fig. 5, the calculated energy gaps for C12-BN and O16-BN are 3.02 and 3.54 eV,respectively,indicating that both C12-BN and O16-BN are insulators. Interestingly, the insulating character of C12-BN is different from the semiconducting property of BC12 carbon,which has the structure similar to C12-BN.Also,O16-BN andbco-C16 carbon, with similar crystal structures, show different electronic properties. The bco-C16 has been reported to be a topological node-line semimetal before. Moreover,from the DOSs of C12-BN and O16-BN, it is seen that the conduction band minimum(CBM)is mainly originated from the B atoms, while the valence band maximum(VBM)is mostly contributed by the N atoms.
To provide more characterization and information for further experimental observation in future, the x-ray diffraction(XRD) spectra are calculated for C12-BN and O16-BN with a wavelength of 1.54,together with those of existing c-BN,h-BN and BC8-BN phases. As shown in Fig. 6, each phase has its own characteristic peaks. Specifically,the characteristic peaks of c-BN are located at 2θ=43.4°and 74.3°,consistent with both experimental values and other simulated results.The strongest peak of h-BN is located at 2θ=26.7°.For BC8-BN, four main XRD peaks are located at 39.7°, 44.6°, 49.0°and 78.8°, respectively. The most intense peak of C12-BN is located at 2θ=42.3°,together with other characteristic peaks at 49.3°, 66.8°and 82.5°, respectively. However, there are much more peaks for O16-BN,i.e.,29.6°,32.9°,40.1°,42.9°,43.7°,45.1°and 47.6°. The simulated XRD spectra predicted here are expected to be helpful in distinguishing C12-BN and O16-BN phases in future experiments.
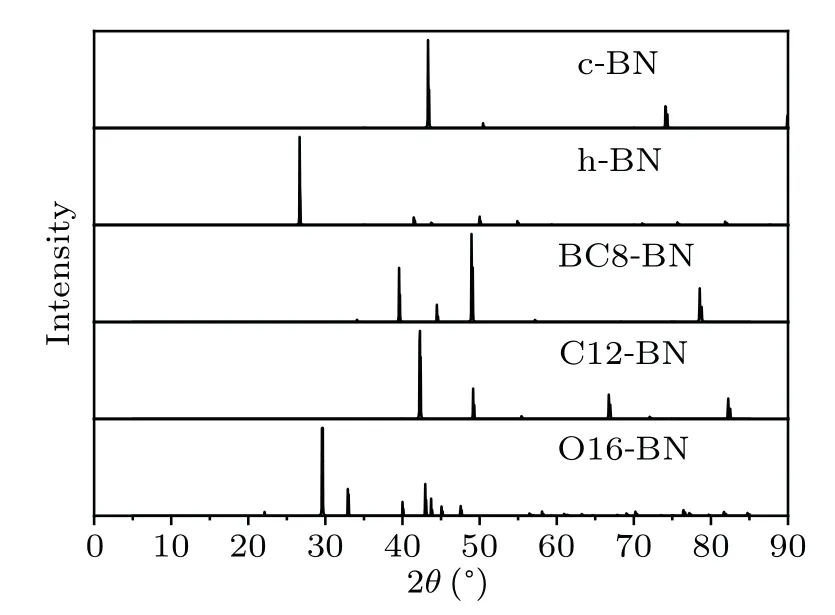
Fig.6. Simulated XRD spectra of c-BN,h-BN,BC8-BN,C12-BN and O16-BN.The x-ray wavelength is set as 1.54 .
To synthesize this new phase of C12-BN experimentally,it is interesting to note that C12-BN can be viewed as a polymerized form of BN nanotubes, as shown in Fig.1(c), which is similar to the case of BC12 carbon that can be viewed as a polymerized form of(3,0)carbon nanotube. The structural relations suggest that C12-BN may be synthesized by the polymerization of selected BN nanotubes. For the synthesis of O16-BN, it is worth referring to a recently reported Cco-C8carbon,[41]which can be obtained by (4,4) carbon nanotube under nonhydrostatic pressure. Since borazine(B3N3H6)has been recently used in the synthesis of BN nanotubes and BN nanosheets,[42]we anticipate that borazine will be a potential starting material to synthesize both C12-BN and O16-BN.Moreover,the synthetic procedure for molecular structures has been recently reported by Aymeet al.[43]
4. Conclusion
In summary, we have predicted two new BN allotropes C12-BN and O16-BN by means of first-principles calculations. C12-BN is a cubic BN structure inI¯43dsymmetry containing purely sp3hybridized six-membered rings. O16-BN,belonging toIma2 space group,is an orthorhombic phase containing only sp2bonds. Based on the results of total energy and cohesive energies,the energetic stabilities of both C12-BN and O16-BN are confirmed as well as their dynamical stabilities,since no imaginary phonon frequencies appear throughout the whole Brillouin zone.Molecular dynamics simulations are carried out to verify the thermal stabilities of C12-BN and O16-BN.Further electronic band structure calculations reveal that C12-BN and O16-BN are insulators with direct band gaps of 3.02 and 3.54 eV,respectively. The C12-BN structure possesses excellent bulk modulus and Vickers hardness, making it a potential incompressible and superhard material. The mechanical properties of O16-BN are relatively poor. However,O16-BN has low density and porous structure, which leads to potential applications in adsorption materials, e.g., hydrogen storage. The XRD spectra of C12-BN and O16-BN are also provided for possible experimental observation. Therefore,our findings not only enrich the existing allotropes family of BN-based material,but also stimulate further experimental research.
Acknowledgement
Project supported by PhD research startup foundation of Civil Aviation University of China(Grant No.2020KYQD94).
猜你喜欢
杂志排行
Chinese Physics B的其它文章
- A broadband self-powered UV photodetector of a β-Ga2O3/γ-CuI p-n junction
- High-sensitive terahertz detection by parametric up-conversion using nanosecond pulsed laser
- High efficiency,small size,and large bandwidth vertical interlayer waveguide coupler
- High-fidelity resonant tunneling passage in three-waveguide system
- An analytical model for cross-Kerr nonlinearity in a four-level N-type atomic system with Doppler broadening
- Determine the physical mechanism and source region of beat wave modulation by changing the frequency of high-frequency waves