Effect and mechanism of knockout RhoE on the protein expression profiles in cardiac tissues of diabetic rats based on 4D proteomics
2022-02-17ZHAOYunSHIKaijiaWUCaixiaZOUYuanWULinxuCHENXuSHENZhihuaGUOJunliJIEWei
ZHAO Yun, SHI Kai-jia, WU Cai-xia, ZOU Yuan, WU Lin-xu, CHEN Xu, SHEN Zhihua, GUO Jun-li✉, JIE Wei✉
1. Hainan Provincial Key Laboratory of Tropical Cardiovascular Disease Research, the First Affiliate Hospital, Hainan Medical University, Haikou 571199, China
2. Department of Pathology, School of Basic Medicine, Guangdong Medical University, Zhanjiang 524023, China
3. Department of Endocrinology, the First Affiliated Hospital, Hainan Medical University, Haikou 571199, China
Keywords:
ABSTRACT Objective: To analyze the effect of RhoE gene expression change on protein expression profiles in the cardiac tissue of diabetic rats, and analyse the possible underlying regulatory mechanism. Methods: Six-week-old male RhoE gene knockout (KO) and wild-type (WT)Sprague Dawley rats were injected intraperitoneally with streptozotocin (70 mg/kg) to establish the type 1 diabetes model (T1DM), with injection of the same amount of sodium citrate saline as the control group. A week later, the fasting blood glucose of the rats was measured daily, and blood glucose concentration 16.7 mmol/L was regarded as a successful model. Two additional weeks later, the hearts of the rats in each group were removed and fourdimensional label-free quantitative proteomics technology (4D-LFQ) was used to analyze the changes of protein profiles in the heart tissue. The related functions, enrichment signals,and protein-protein interaction network (PPI) of the differentially expressed proteins were analyzed. Results: T1DM rat models were successfully established. Taking a fold change> 1.5 and P < 0.05 as the threshold, a total of 2 931 quantifiable proteins were identified.In the non-diabetic state, the KO group had 26 and 45 significantly up- and downregulated proteins, respectively, compared with the WT group; in the diabetic state, the KO group had 19 and 28 significantly up- and downregulated proteins, respectively, compared with the WT group. The GO annotation results showed that most of the differentially expressed proteins were located in the extracellular matrix, and their biological functions were mainly concentrated in the immune response and energy metabolism. KEGG analysis showed that the signaling pathways associated with the differentially expressed proteins in cardiac tissue after RhoE knockout were mainly related to ribosomes and fat digestion and absorption. Protein interaction network analysis showed that in the cardiac tissue of the KO group, there were more Col1α1- and Col1α2-interacting proteins among the upregulated proteins, and among the down-regulated proteins, related proteins involved in the ribosomal pathway interact more in the network. Conclusion: RhoE knockout significantly changes the protein expression profiles in diabetic cardiac tissue, affecting multiple signaling pathways closely related to diabetic cardiomyopathy. The results provide insights into the pathogenesis and therapeutic target screening of diabetic cardiomyopathy.
1. Introduction
Diabetic cardiomyopathy (DCM) is a myocardial disease complicated by diabetes that cannot be explained by hypertensive heart disease, coronary atherosclerotic heart disease, and other heart diseases of known etiology [1, 2]. The complex pathogenesis has not yet been elucidated. Therefore, it is extremely important to find suitable biomarkers for the prevention and treatment of DCM. The recent development of transcriptomics [3], proteomics [4],metabolomics [5], modification omics [6], and other studies provide potential solutions for this.
RhoE, also known as RND3, is a member of the Rho GTPase protein superfamily. RhoE has diverse roles and is related to biological functions such as cell proliferation, the cell cycle,apoptosis, migration, and differentiation [7]. It has been reported that abnormal expression of RhoE is associated with cardiovascular diseases [8-10]. Our laboratory previously constructed the RhoE gene stably knocked out H9C2 cardiomyocyte cell line based on CRISPR/Cas9 technology, screened differentially expressed genes, and enriched the RhoE-related signaling pathways by microarray. We found that RhoE is involved in the regulation of pathways including the cholesterol biosynthesis pathway, oncostatin-M signaling,interferon signaling, and TGF-β1 signaling, highlighting the important role of RhoE in cardiac disease [11]. So far, however, there have been no reports on the relationship between RhoE and DCM.As a high throughput screening method for differentially expressed proteins, 4D proteomics has recently received increased attention [12,13]. This study intends to use 4D non-label quantitative proteomics technology to detect the protein expression profiles of RhoE expression changes in the heart tissue of diabetic rats, characterize the differentially expressed proteins regulated in the heart tissue,and combine bioinformatics methods to explore the differentially expressed proteins. The involved functions, distribution, and related signaling pathways will provide ideas and methods for deciphering the role of RhoE in DCM.
2. Materials and methods
2.1 Main reagents and instruments
Main reagents: streptozotocin (YEASEN, China), dithiothreitol,trifluoroacetic acid, chloroform, urea, tetramethylethylenediamine,Seppro® Rat Spin Columns (Sigma-Aldrich, USA), acetonitrile,methanol, TMT labeling reagent, protein marker (ThermoFisher Scientific, USA), PAGE silver staining kit, DNA extraction phenol reagent (Beijing Solarbio), Pierce Top 12 Abundant Protein Depletion Spin Columns, nitrocellulose filter membrane, and polyvinylidene fluoride ethylene (Bio-Rad Laboratories, USA).
Main instruments: NanoElute, high-resolution mass spectrometer Bruker timsTOF Pro, ultrasonic instrument (ThermoFisher Scientific, USA), electrophoresis instrument, electrophoresis tank,membrane transfer tank (Bio-Rad, USA).
2.2 Experimental animals and groups
Adult heterozygous Sprague Dawley (SD) rats with a systemic knockout of RhoE (RhoE+/-, male, weight 200-250 g) and wildtype SD rats (male, 6 weeks old, weight 200-250 g) were provided by Suzhou Cyagen Biotechnology Co., Ltd. and were raised in the Laboratory Animal Center of Guangdong Medical University for breeding and genotyping [14]. A diabetic rat model was established by combining a high-fat diet with one-time streptozotocin (STZ,70 mg/kg). After 2 weeks of streptozotocin injection, the fasting blood glucose value was16.7 mmol/L three consecutive times.Grouped cardiac tissue was used for the proteomic studies. The groups were as follows: normal control rats (WT_NG), WT rats were intraperitoneally injected with a volume of sodium citrate saline equal to the streptozotocin treatment; diabetic rats (WT_HG),WT rats were induced to type 1 diabetes (T1DM) by intraperitoneal injection of streptozotocin; RhoE knockout control rats (KO_NG), RhoE+/-rats with intraperitoneal injection of sodium citrate saline; RhoE knockout diabetic rats (KO_HG), RhoE+/-rats with intraperitoneal injection of streptozotocin to induce T1DM. Animals were raised in an specific pathogen free environment, the diabetes group was given a high-fat diet and regular drinking water, and the control group was given a regular diet and drinking water.All experimental operations were in compliance with the ethical requirements for experimental animals of Guangdong Medical University (ethics number: GDY2016003). There were three rats in each group (n = 3).
2.3 Protein extraction and enzymatic hydrolysis
Refer to the methods of others [15, 16]. Briefly, a mortar was precooled with liquid nitrogen and then place an appropriate amount of myocardial sample. After adding liquid nitrogen, the tissue pieces were completely ground into powder. Four times the powder volume of lysis buffer (containing 1% protease inhibitor, 50 mM nicotinamide, 3 μM sirtuin inhibitor, and 8 M urea) was added to each group of samples for sonication. After centrifugation (4 ℃, 12 000 g, 10 min), the supernatant was transferred to another sterile centrifuge tube, and the protein concentration was determined using a BCA kit. An equal amount of each sample protein was taken, and the volume of each group was adjusted to the same volume with lysis buffer. Gently, 20% trichloroacetic acid was added, vortexed,and mixed well, and then precipitated at 4 ℃ for 2 h, centrifuged,and the precipitate was washed three times with cold acetone. The precipitate was placed on an ultra-clean table to dry, and 200 mM tetramethylammonium bromide was added and then dispersed by ultrasound. Trypsin was added at a ratio of protease:protein = 1:50,and enzymolysis was carried out overnight. Dithiothreitol was added to adjust the final concentration to 5 mM, and the reaction was performed at 56 ℃ for 30 min. Finally, iodoacetamide was added to adjust the final concentration to 11 mM. Each sample was incubated in the dark for 15 min before use.
2.4 LC-MS analysis and data analysis
Chromatographic mass spectrometry analysis was completed using the technology platform of Hangzhou PTMBIO Company, and the relevant experimental steps are reported in other studies [15-17].Briefly, mobile phase A was prepared by mixing an aqueous solution containing 0.1% formic acid and 2% acetonitrile, and mobile phase B was prepared by mixing a solution containing 0.1% formic acid and 100% acetonitrile. The peptide fragments of each sample were dissolved in mobile phase A and separated by the Nano Elute ultrahigh performance liquid phase system. The liquid phase gradient was set as 0-70 min, 6%-24% B; 70-84 min, 24%-32% B; 84-87 min,32%-80% B; and 87-90 min, 80% B. The flow rate was maintained at 450 nL/min. After the peptides were separated by the ultra-high performance liquid phase system, the capillary ion source was added for ionization, and then the ionized peptides were analyzed by timsTOF Pro mass spectrometry (MS). The scan interval of the secondary MS was set to 100-1700. The data acquisition mode was set to Parallel Accumulation Serial Fragmentation (PASEF) mode.Using Maxquant (v1.6.15.0) to search the MS data, a total of 29,940 sequences were found in Rattus_norvegicus_10116_PR_20201214.fasta. The false positive rate (FDR) caused by random matching was calculated after adding the reverse library, and the false discovery rate of proteins and peptides was adjusted to 1%. After the database search was completed, a quality control series was required to ensure that the quality of the results met standards. Significantly upregulated expression was defined as a differential expression level change (FC)> 1.5 and P < 0.05, and significantly downregulated expression was defined as FC < 1.5 and P < 0.05.
2.5 Bioinformatics analysis
The differentially expressed proteins in each comparison group were subjected to Gene Oncology (GO) classification and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis, and Fisher’s exact test was used to evaluate the functional classification of the differentially expressed proteins and the significance level of the pathway. A hierarchical clustering method was applied to draw a cluster heatmap. The differentially expressed protein database numbers or protein sequences in different comparison groups were compared with the STRING (v.11.0)protein network interaction database, and the differential protein interaction relationship was extracted according to a confidence score > 0.7.
3. Results
3.1 Protein identification and analysis
After enzymatic hydrolysis of rat hearts from each group,the protein was quantitatively analyzed by BCA and met the requirements of mass spectrometry analysis. Through liquid chromatography-mass spectrometry analysis and a database search,a total of 2,067,933 MS spectra were obtained. These consisted of 308,807 spectra matching the theoretical MS spectra, 29,739 peptides in total, 26,946 unique peptides in total, and 3597 proteins that were identified. Of these, 2931 proteins were quantified. The length of the peptide segment was mainly 7-20 amino acids, most of the proteins corresponded to more than two peptide segments,the coverage of most proteins was below 20%, and the molecular weights of the proteins were mainly distributed between 10-120 KD. A heat map was drawn by calculating the Pearson Correlation Coefficient between all samples. The correlation between the corresponding peptides, protein distribution, and samples is shown in Figure 1.
3.2 Differential protein screening in cardiac tissue before and after RhoE knockout
Taking FC > 1.5 and P < 0.05 as the threshold for significant upregulation, and FC<1.5 and P<0.05 as the standard for significant downregulation, it was found that, compared with the WT_NG group, the WT_HG group upregulated 198 and downregulated 331 differentially expressed proteins (Fig 2A). The KO_NG group upregulated 26 and downregulated 45 differentially expressed proteins (Fig 2B). Compared with KO_NG, KO_HG upregulated 173 and downregulated 255 proteins (Fig 2C). Compared with WT_HG, KO_HG upregulated 19 and downregulated 28 differentially expressed proteins (Fig 2D). The corresponding top ten proteins with the most significant expression are shown in Table 1.
3.3 Annotation of the subcellular localization of differential proteins in cardiac tissue before and after RhoE knockout

Fig 1 Protein identification and analysis
Compared with the WT_NG group, the upregulated proteins in the heart tissue of the KO_NG group were mainly located in mitochondria, the extracellular matrix, and the nucleus, while the downregulated proteins were mainly distributed in the cytoplasm,extracellular matrix, and nucleus. The upregulated proteins in the heart tissue of the WT_HG group were mainly located in the cytoplasm, mitochondria, and extracellular matrix, while the downregulated proteins were mainly distributed in the cytoplasm,nucleus, and extracellular matrix. Compared with the WT_HG group, the upregulated proteins in the heart tissue of the KO_HG group were mainly located in the extracellular matrix, cytoplasm,and mitochondria, and the downregulated proteins were mainly distributed in the cytoplasm, nucleus, and extracellular matrix.Compared with the KO_NG group, the upregulated proteins in the heart tissue of the KO_HG group were mainly located in the cytoplasm, extracellular matrix, and mitochondria, and the downregulated proteins were mainly distributed in the cytoplasm,mitochondria, and nucleus. The subcellular localization annotation classification map of the differential proteins compared between the groups is shown in Figure 3.
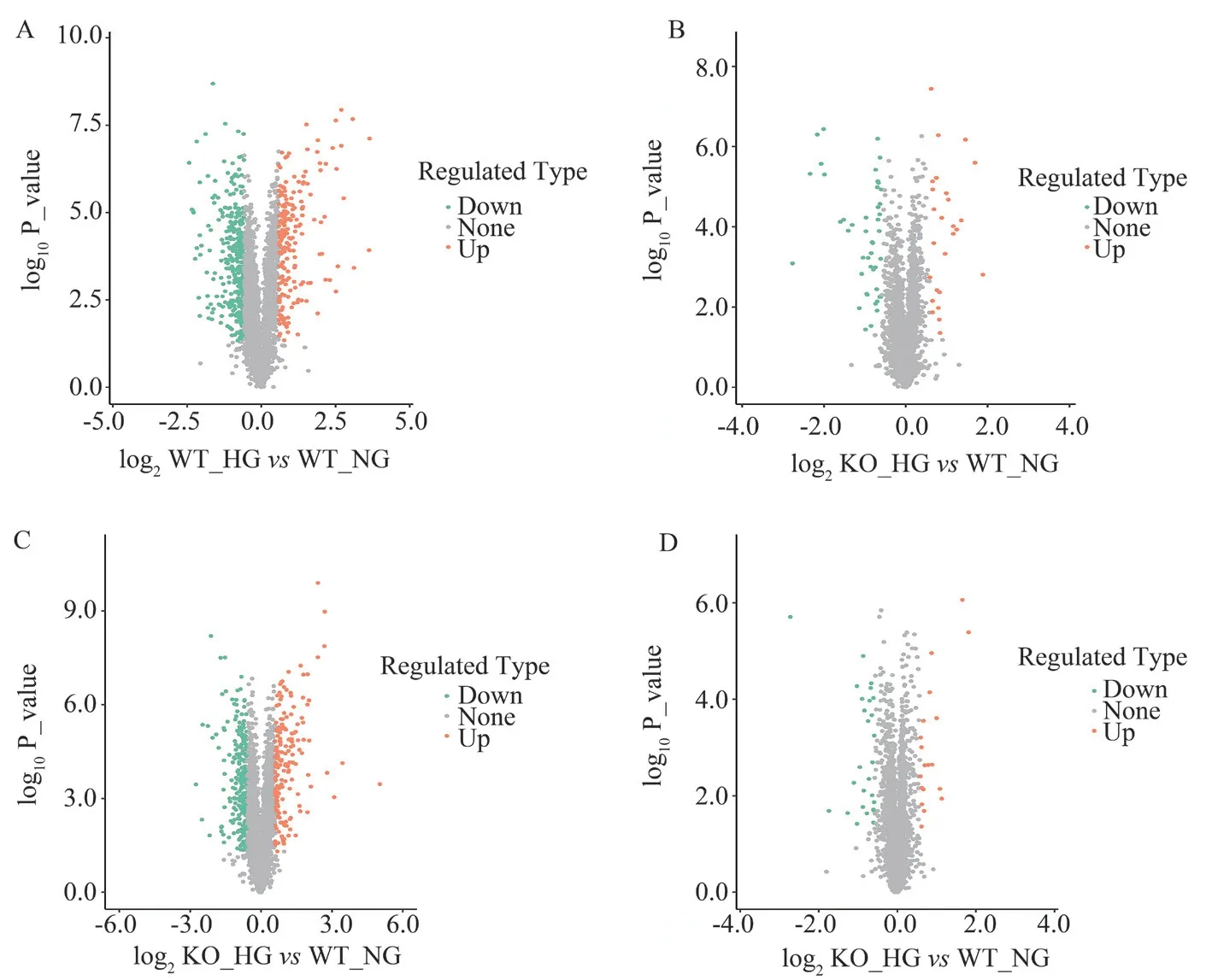
Fig 2 Volcano plot of differentially expressed proteins in cardiac tissue before and after RhoE knockout
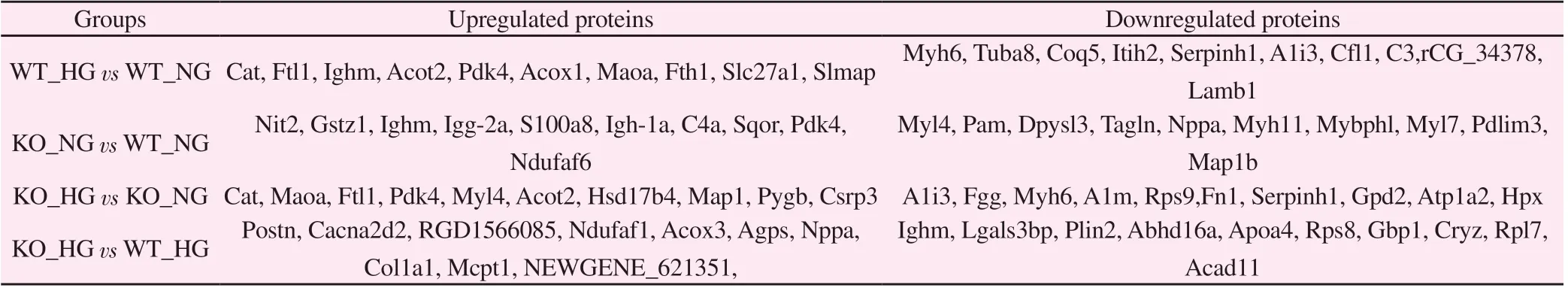
Tab 1 List of top ten differentially expressed proteins
3.4 GO annotation of differentially expressed proteins
GO annotation was performed on the screened differential proteins for the three levels of cell composition, molecular function, and biological process. The results showed that, compared with WT_NG,the upregulated proteins in the heart tissue of the WT_HG group were mainly distributed in peroxisomes, and the downregulated proteins were mainly distributed in ribosomal subunits. The molecular functions of the upregulated proteins were mainly concentrated in cofactor binding, and the molecular functions of the downregulated proteins were mainly concentrated in the structural components of ribosomes. The upregulated proteins were mainly involved in the biological process of cellular lipid metabolism, and the downregulated proteins were mainly involved in the peptide biosynthesis process (Fig. 4A, B). Compared with WT_NG, the upregulated proteins in the heart tissue of the KO_NG group were mainly distributed in the immunoglobulin complex, and the downregulated proteins were mainly distributed in the extracellular region. The molecular functions of the upregulated proteins were mainly concentrated in the binding of immunoglobulin receptors,and the downregulated proteins were mainly concentrated in the extracellular matrix structure constituent. The upregulated proteins were mainly involved in the biological process of the immunoglobulin-mediated immune response, and the downregulated proteins were mainly involved in the biosynthesis of aminoglycans(Fig. 4C, D). Compared with KO_NG, in the heart tissue of the KO_HG group, the upregulated proteins were mainly distributed in the peroxidase matrix, and the downregulated proteins were mainly distributed in ribosomal subunits. The molecular functions of the upregulated proteins were mainly concentrated in cofactor binding, and the downregulated proteins were mainly concentrated in ribosome structural components. The upregulated proteins were mainly involved in the biological process of peroxidase composition,and the downregulated proteins were mainly involved in the process of peptide biosynthesis (Fig 4E, F). Compared with WT_HG, the upregulated proteins in the heart tissue of the KO_HG group were mainly distributed in the extracellular space, and the downregulated proteins were mainly distributed in the cytoplasmic large ribosomal subunit. The molecular functions were mainly concentrated in the structural components of the ribosome; the upregulated proteins were mainly involved in the biological process of blood pressure regulation, and the downregulated proteins were mainly involved in the biological process of neutral lipid decomposition (Fig 4G, H).
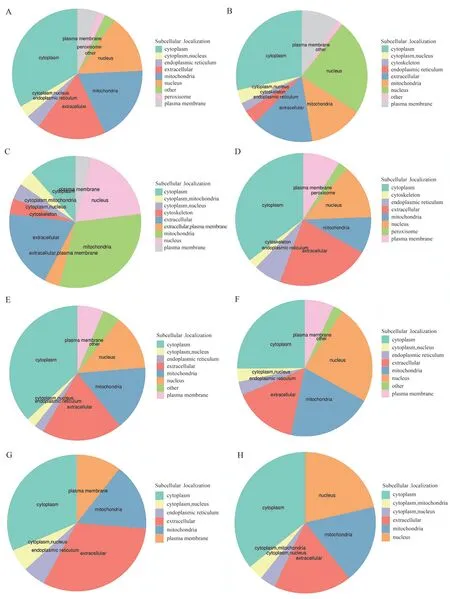
Fig 3 Annotation of subcellular localization of differential proteins in cardiac tissues before and after RhoE knockout

Fig 4 GO annotation of differentially expressed proteins
3.5 KEGG pathway enrichment analysis of differentially expressed proteins
After comparing the differentially expressed proteins with the KEGG database, the differential signal analysis showed that,compared with the WT_NG group, the upregulated signaling pathways in the heart tissue of the WT_HG group were mainly peroxisomes, the effect of cytochrome P450 on the metabolism of exogenous organisms, and arachidonic acid metabolism (Fig 5A). The downregulated signaling pathways mainly included ribosomes, coronavirus disease COVID-19, and protein digestion and absorption (Fig 5B). Compared with the WT_NG group, the upregulated signaling pathway in the heart tissue of the KO_NG group was the rno04621 nod-like receptor signaling pathway (Fig 5C), and the downregulated signaling pathways were mainly protein digestion and absorption, vascular smooth muscle contraction, and the relaxin signaling pathway (Fig 5D). Compared with KO_NG,the upregulated signaling pathways in the heart tissue of the KO_HG group were mainly terpenoid backbone biosynthesis, arachidonic acid metabolism, and peroxisomes (Fig 5E), and the downregulated signaling pathways were mainly ribosomes, Staphylococcus aureus infection, and coronavirus disease COVID-19 (Figure 5F). Compared with the WT_HG group, the upregulated signaling pathways involved in the differentially expressed proteins in the heart tissue of the KO_HG group were mainly protein digestion and absorption,the interaction of extracellular matrix receptors, and AGE-RAGE signaling in diabetic complications, and the downregulated signaling pathways were mainly vitamin digestion and absorption, fat digestion and absorption, and cholesterol metabolism (Fig. 5G).
3.6 Protein interaction network analysis
The STRING protein network interaction database (V.11.0) showed that two of the upregulated proteins in the KO_HG group compared with the WT_HG group, Col1 1 and Col1 2, interacted with each other in cardiac tissue. and the related proteins involved in the ribosomal pathway (Rps8, Rpl7, Rpl18, Rpl23, and Rpl31) among the downregulated proteins have an increased number of interacting proteins in the network (Figure 6).

Fig 5 Bubble chart of KEGG pathway enrichment analysis of differentially expressed protein
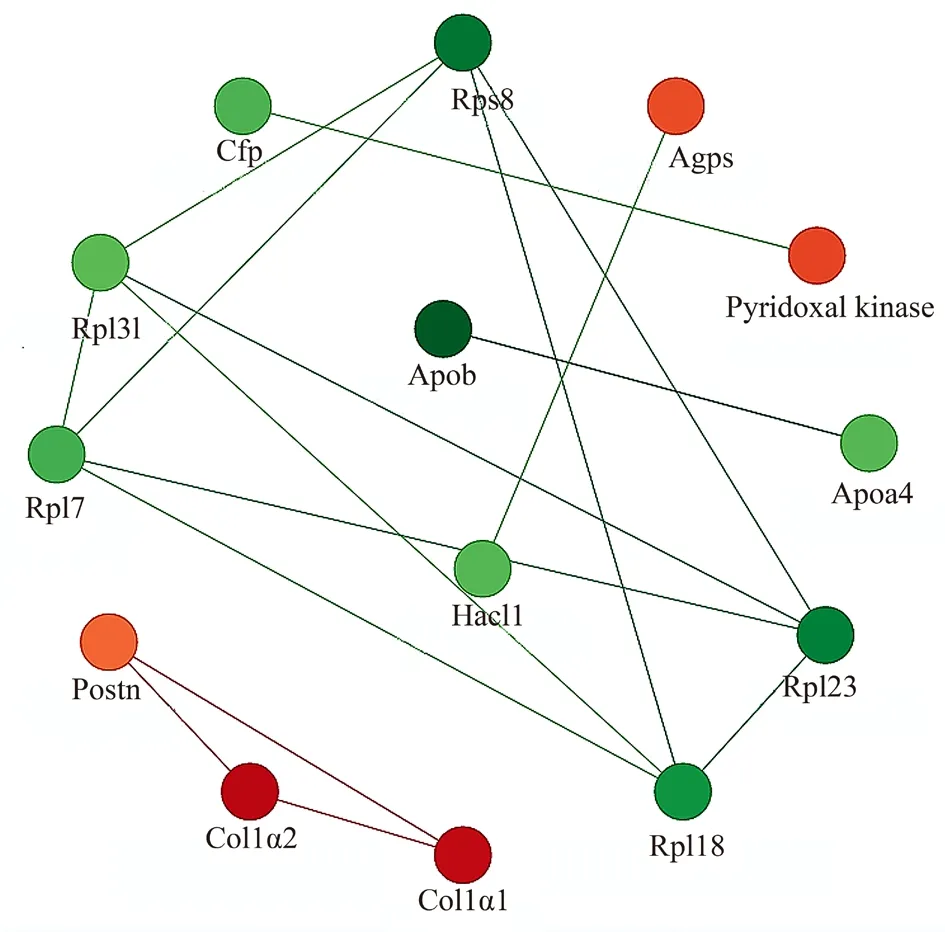
Fig 6 Analysis of protein interaction network
4. Discussion
The pathogenesis of DCM is complex. To understand the role of RhoE in DCM, this study replicated a diabetes model using RhoE gene edited model animals and used the latest 4D proteomics to study the relationship between RhoE and DCM from a global perspective.
In the past, the use of RhoE gene edited cells and animal models has revealed many new functions of RhoE in cardiovascular diseases[8-11]. In this study, Suzhou Cyagen Biotechnology Company was commissioned to construct a RhoE gene knockout heterozygous SD rat based on CRISPR/Cas9 genome editing technology. The rats were further bred and identified to obtain the experimental animals required for the experiment, and then the classic intraperitoneal injection of streptozotocin was used to induce acute type 1 diabetes.The successful replication of this model laid the foundation for subsequent research. Then, the rat hearts of each group were isolated,and the protein lysates required for proteomics were obtained by traditional enzymatic hydrolysis. Through liquid chromatographymass spectrometry, a total of 2,067,933 MS spectra, 29,739 peptides,3597 proteins, and 2931 quantifiable proteins were isolated. Through strict quality control, the peptide length, protein coverage, and protein molecular weight in this group all met the quality control requirements. The correlation analysis showed that the Pearson Correlation Coefficient of the samples within the group was greater than 0.99, and the Pearson correlation coefficient of the samples between the groups was between 0.95 and 0.97. This indicated that the animals used in this study had little intra-group difference and good repeatability but there were significant differences between groups, and suggested that the animals in the test samples met the experimental requirements.
Among the 3597 proteins identified, it was found that, compared with the WT_NG group, 198 differentially expressed proteins were upregulated and 331 were downregulated in the WT_HG group.Representative proteins include Cat, Ftl1, Ighm, Acot2, Pdk4, Myh6,Tuba8, Coq5, Itih2, and Serpinh1. The KO_NG group upregulated 26 differentially expressed proteins and downregulated 45, and representative proteins include Nit2, Gstz1, Ighm, Igg-2a, S100a8,Myl4, Tagln, Myh11, Mybphl, and Myl7. Compared with KO_NG,KO_HG upregulated 173 proteins and downregulated 255 proteins,and representative proteins include Cat, Maoa, Ftl1, Pdk4, Myl4,A1i3, Fgg, Myh6, A1m, and Rps9. Compared with WT_HG, KO_HG significantly upregulated 19 differentially expressed proteins and significantly downregulated 28, and representative proteins include Postn, Cacna2d2, Ndufaf1, Col1a1, Col1a2, Ighm, Lgals3bp, Plin2,Abhd16a, and Apoa4. Subcellular structure localization analysis showed that, in the non-diabetic state, the most obvious changes in protein distribution before and after RhoE gene knockout were in the cytoskeleton, peroxisome, and nucleus; early studies suggested that RhoE regulates the cytoskeleton [7]. The results of this study also suggest that RhoE knockout significantly altered the expression of cytoskeletal proteins. In addition, the changes to the peroxisome before and after RhoE knockout also suggest that the peroxisome is related to RhoE and oxidative stress, consistent with an earlier report [18]. Changes in nuclear protein distribution suggest that RhoE is involved in the regulation of gene expression. Interestingly, the most significantly altered distributions in the diabetic state were in the plasma membrane and extracellular regions. Our study showed that downregulation of RhoE expression promoted the secretion of collagen Col1a1 and Col1a2 in diabetic rats, which was closely related to the altered distribution of extracellular regions.
GO annotation analysis of differentially expressed proteins showed that the main components of the extracellular space and related platelet-derived growth factor binding functions, and the biological processes of blood pressure upregulation were significantly upregulated in RhoE-knockout diabetic rats, while the related components of the cytoplasmic large ribose subunit and the structure and function of ribosomes, and the breakdown of neutral lipids were significantly downregulated. This is consistent with previous reports of RhoE and cardiovascular related studies [10]. Through KEGG analysis, this study found that the signaling pathways involved in the differentially expressed proteins in the cardiac tissue of the KO_NG group compared with the WT_NG group were mainly the relaxin signaling pathway, focal adhesion, and the PI3K/Akt signaling pathway. The signaling pathways involved in the differentially expressed proteins in the heart tissue of the KO_HG group compared with the WT_HG group were mainly ribosomes, coronavirus disease COVID-19, and fat digestion and absorption. All the above suggest that RhoE may participate in the pathophysiological process of DCM through these signaling pathways. Among them, the PI3KAkt signaling pathway is crucial in DCM. Inhibition of autophagy in PI3K/Akt pathway can improve insulin resistance [19]. Upregulation of microRNA-203 can target PIK3CA through inactivation of the PI3K/Akt signaling pathway, thus providing a good solution for DCM [20]. In addition, H2 and H3 relaxin inhibit high-glucoseinduced apoptosis of neonatal rat ventricular myocytes [21], and the highly expressed ribosomal protein (RPS4Y1) in endothelial cells may lead to high-glucose-induced dysfunction by regulating the p38 MAPK signaling pathway [22]. In addition, lipid metabolism disorders also play a critical role in the development of DCM,and correcting lipid metabolism disorders can alleviate cardiac insufficiency in diabetic mice [23]. The loss of RhoE will further lead to changes in the activation of the above signaling pathways,thereby affecting the occurrence and development of DCM.This provides a possible direction for further exploration of the molecular mechanism of the pathogenesis of DCM in the later stage.Hospitalized SARS-CoV-2-infected patients with type 2 diabetes develop acute cardiovascular syndrome, while the underlying mechanisms associated with acute cardiac injury remain unclear[24]. In this study, the coronavirus disease COVID-19 pathway was enriched in the cardiac tissue of RhoE-deficient diabetic rats, which seems to provide new ideas for the study of the mechanism of acute cardiac injury in patients with diabetes infected by COVID-19.
In the protein interaction network analysis, we found that the upregulated proteins in the KO_HG group compared with the WT_HG group included more Col1 1- and Col1 2-interacting proteins.Among the downregulated proteins, related proteins involved in the ribosomal pathway were present in the network. There were more interacting proteins in the KO_NG group compared with the WT_NG group, and we also get the interacting proteins (unpublished data). These data provide a more intuitive experimental basis for elucidating the pathogenesis of DCM.
This study has certain limitations. First, the research object of this experiment is RhoE-knockout rats, not a heart-specific knockout, so this may have an impact on the results. Second, this study used rat hearts as a model, and the applicability of our conclusions to human research requires further verification. Finally, the enriched related proteins and KEGG signals are still being verified.
In conclusion, this study applies the latest 4D-LQF proteomic technology to reveal the changes of protein expression profiles in diabetic heart tissue after loss of RhoE expression for the first time,the biological functions related to differentially expressed proteins were preliminarily analyzed, and the main signaling pathways involved in differentially expressed proteins were enriched. The results of this study provide an experimental reference for the clinical diagnosis and treatment of DCM from a macro perspective.
Conflicts of interest: All authors declare no conflict of interest.
Author Contributions: Thesis design: JIE Wei, GUO Jun-li; Animal experiments and sample preparation: WU Cai-xia, ZOU Yuan; Data statistics, graph production and literature search: ZHAO Yun, SHI Kai-jia, WU Lin-xu, CHEN Xu; Manuscript writing: ZHAO Yun,SHI Kai-jia, SHEN Zhi-hua, GUO Jun-li, JIE Wei; Fund acquisition:JIE Wei.
杂志排行

Journal of Hainan Medical College的其它文章
- Research progress on pathogenesis of ulcerative colitis
- Advances in the application of optical coherence tomography in the assessment of ischemic stroke
- Meta-analysis of the clinical efficacy of Chinese herbal decoction combined with arthroscopy in the treatment of gouty arthritis
- Study on syndrome distribution and medication characteristics of patients with rectal cancer in the real world
- Experimental study of TGF-β1/Smads pathway inhibition of macrophage polarization based on miR145-5P negative feedback regulation
- Exploring the key pathways of tetrandrine in the treatment of early silicosis based on bioinformatics and in vitro experiments