QuEChERS-超高效液相色谱-串联质谱法检测茶叶中胺菊酯
2022-02-16叶美君杜颖颖李文萃陆小磊刘相真
叶美君,杜颖颖,李文萃,陆小磊,刘相真
(中华全国供销合作总社杭州茶叶研究所,浙江 杭州 310016)
0 引言
胺菊酯CAS号为7696-12-0,分子量为331.4,纯品为白色结晶固体,具有除虫菊气味,在弱酸性条件下稳定。在30℃条件下,水中溶解度为4.6 mg/L;在25℃条件下溶于苯、二甲苯、甲苯和丙酮,微溶于甲醇和乙醇。胺菊酯为世界卫生组织推荐用于公共卫生的主要杀虫剂之一,用于防治蚊、蝇、蟑螂等卫生害虫,具有触杀作用、对蜚蠊有驱赶作用,但致死性能差,有复苏现象。胺菊酯属于神经毒剂,大量摄入会致人中毒,引起头痛、头昏、恶心呕吐、双手颤抖,重者抽搐或惊厥、昏迷、休克[1-2]。目前,国内胺菊酯的研究主要集中在原药制备、剂型分析,食品中胺菊酯的检测方法研究较少,相关检测方法主要集中在气相色谱或气相色谱-质谱法,液相色谱或液相色谱-质谱法检测胺菊酯相关报道较少[3-5]。
在茶园种植过程中尚未报道胺菊酯作为杀虫剂使用,茶叶中胺菊酯等污染物质可能从生产和流通过程中引入。监测茶叶中胺菊酯残留水平和排查该类物质污染源的首要任务是建立一种快速、灵敏和准确的检测方法。鉴于此,选择高灵敏度的UPLCMS/MS作为检测仪器,采用GCB、PSA、C18和NH2等填料为吸附剂对茶叶提取液进行净化,降低UPLC-MS/MS检测过程中基质干扰,同时该方法具有准确、灵敏和通用性强等优点,可为茶叶安全风险监测与评估提供有效的技术支撑。
1 材料与方法
1.1 仪器与设备
UPLC/TSQ Quantum Access MAX超高效液相色谱-三重四极杆质谱联用仪,美国Thermo Fisher Scientific公司产品;分析天平(感量0.000 1 g),瑞士METTLER TOLEDO公司产品;SC-3610型低速离心机,安徽中科中佳科学仪器有限公司产品;VORTEX-5型旋涡混合器,其林贝尔Kylin-bell产品;HY-5型回旋振荡器,江苏省金坛市荣华仪器有限公司产品;CS501旋转蒸发仪,上海瑞立科学仪器有限公司产品;MTN-2800D型氮吹浓缩装置,天津奥特赛恩斯仪器有限公司产品。
1.2 试剂与材料
胺菊酯(100.2 μg/mL,1 mL,AccuStandard);磷酸三苯酯TPP(分析纯,含量99%),河北百灵威超精细材料有限公司提供;乙腈、甲醇和甲酸(色谱纯),德国Merck公司提供;乙酸铵(色谱纯),美国Fluka公司提供;乙腈和甲苯(分析纯),华东医药股份有限提供;醋酸(分析纯),上海凌峰化学试剂有限公司提供;无水醋酸钠(分析纯),温州吉象化学股份有限公司提供;无水硫酸镁(分析纯),上海试四赫维化工有限公司提供;乙二胺-N-丙基硅烷化硅胶PSA(40~60 μm)、十八烷基硅烷键合硅胶C18(40~60 μm)、石墨化碳黑GCB(40~120 μm)、50 mL塑料螺纹具塞离心管,上海安谱科学仪器有限公司提供;0.5,1,5,10 mL刻度移液管,天津市天玻玻璃仪器有限公司提供;2 mL一次性无菌注射器,华东医药股份有限公司提供;0.22 μm微孔过滤膜(尼龙),上海迪柯马分析技术有限公司提供。茶叶,市售样品;茶粉,茶叶磨碎样品,按照国际标准GB/T 8303—2013制备。
1.3 标准溶液的配制
准确称取0.075 0 g TPP于100 mL容量瓶中,加入乙腈溶解并定容至刻度线,得到750 mg/L的标准储备溶液。取1 mL标准储备溶液,用乙腈配制成0.75 mg/L标准使用溶液。
准确移取1 mL胺菊酯标准物质于10 mL容量瓶中,加入乙腈溶解并定容至刻度线,得到10 mg/L的标准储备溶液。取1 mL标准储备溶液,用乙腈配制成1 mg/L标准使用溶液,取适量1 mg/L标准使用溶液和TPP内标,加入含茶叶基质(阴性样品)的乙腈溶液,配制成2,5,10,50,100 μg/L的基质匹配标准工作溶液,使用内标法定量。
1.4 仪器条件
色谱柱:Thermo Hypersil GOLD C18(150 mm×2.1 mm,1.9 μm);流动相A为水(含5 mmol/L乙酸铵,0.1%(V/V)甲酸),流动相B为甲醇(含5 mmol/L乙酸铵,0.1%(V/V)甲酸);柱温40℃;进样量4.0 μL;电离模式:电喷雾离子化(ESI),电离源极性:正模式,雾化气:氮气,离子喷雾电压3 500 V,雾化室温度120℃,离子传输管温度350℃,碰撞气:氩气,0.2 MPa,扫描模式:SRM多反应监测扫描。
1.5 样品前处理
(1)提取。称取经粉碎的茶叶样2.0 g于50 mL离心管中,加入10 mL水浸泡30 min,然后加入15 mL乙腈(1%醋酸),6 g无水硫酸镁和1.5 g醋酸钠剧烈振荡提取1 min,以转速4 200 r/min离心5 min。
(2)净化。吸取4 mL上清液于内含300 mg无水硫酸镁,300 mg PSA、200 mg C18和200 mg GCB的15 mL塑料离心管中,涡旋混匀1 min,以转速4 200 r/min离心5 min。准确移取1 mL上清液于5 mL塑料离心管中,加入66.67 μL TPP内标,过0.22 μL微孔滤膜,供UPLC-MS/MS检测。
2 结果与分析
2.1 液相色谱-质谱条件优化与选择
2.1.1 质谱条件的优化
将胺菊酯和TPP的标准溶液进入质谱仪器直接分析,采用ESI+模式。在MS+MS/MS模式下,选择信号强度高、稳定性好的准分子离子作为母离子进行优化,超高效液相色谱-串联质谱参数(包括碰撞能量CE、透镜补偿电压Tube lens等)。
胺菊酯的超高效液相色谱-串联质谱参数见表1。

表1 胺菊酯的超高效液相色谱-串联质谱参数
2.1.2 色谱条件的优化
根据胺菊酯的结构特点,采用通用型的C18色谱柱作为分离柱,分别选择乙腈-水、乙腈-甲酸水溶液,在3%~90%有机相质量分数范围内进行梯度洗脱,考查流动相对胺菊酯检测的影响。在正离子模式下,酸性能够促进化合物的离子化,提高化合物的离子化效率和灵敏度,在水相中添加0.1%甲酸时,胺菊酯的灵敏度高,分离效果满意。在水相中添加5 mmoL乙酸铵,胺菊酯峰型对称、信噪比增大、灵敏度提高。
试验结果表明,当选择乙腈-水(含5 mmol/L乙酸铵,0.1%(V/V)甲酸)溶液体系时,胺菊酯的色谱分离、保留和灵敏度情况较为理想,稳定性较好,但内标TPP的稳定性和灵敏度欠佳。选择甲醇-水(含5 mmol/L乙酸铵,0.1%(V/V)甲酸)作为流动相,内标TPP的响应和稳定性显著提升,但胺菊酯的响应相对乙腈溶液略微下降。在有机相B中加入一定量的乙酸铵和甲酸,配置成含5 mmol/L乙酸铵和0.1%(V/V)甲酸的甲醇溶液,保证流动相中甲酸和乙酸铵的浓度一致,以提高样品的稳定性和重复性。综上所述,试验采用水(含5 mmol/L乙酸铵,0.1%(V/V)甲酸)-甲醇(含5 mmol/L乙酸铵,0.1%(V/V)甲酸)作为流动相进行梯度洗脱。
梯度洗脱程序见表2,胺菊酯和TPP标准溶液在LC-MS/MS上的色谱图见图1。

图1 胺菊酯和TPP标准溶液在LC-MS/MS上的色谱图
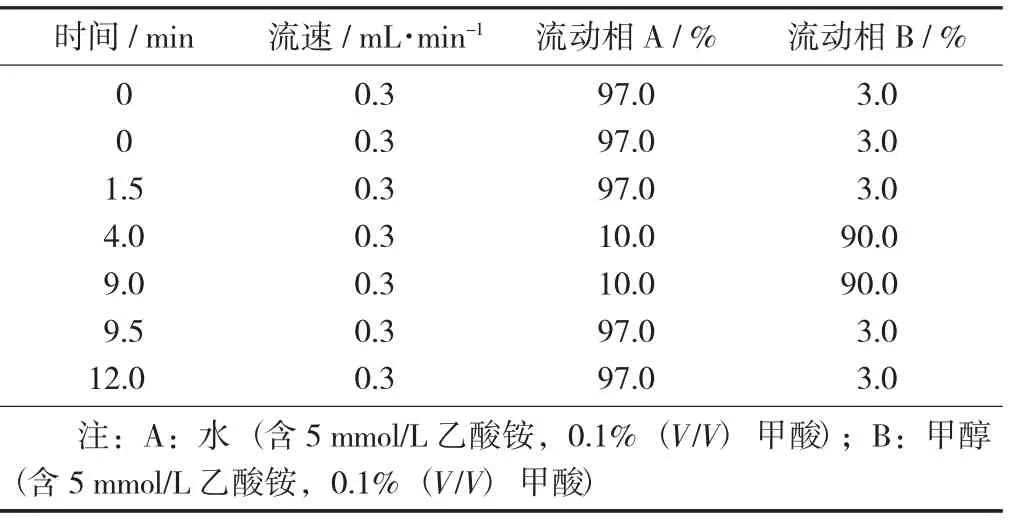
表2 梯度洗脱程序
2.2 前处理方法优化
2.2.1 提取方法的选择
茶叶含水率低,在提取之前加入一定量的水浸泡(非提取剂),基于乙腈良好的提取效率,植物源食品中农药残留检测大量采用乙腈为提取剂[6-8]。前期试验表明,提取剂中加入1%醋酸可改善胺菊酯在UPLC-MS/MS上的峰型,提高检测灵敏度。
提取方式主要有超声、旋涡、振荡和匀浆等,根据前期试验,结合GB 23200.121《食品安全国家标准植物源性食品中331种农药及其代谢物残留量的测定 液相色谱-质谱联用法》中7.3种茶叶和香辛料的前处理方法,采用振荡提取方式。
2.2.2 净化材料的选择与优化
茶叶中有机酸、脂肪和磷脂较高,GCB、PSA、C18和NH2等填料去杂质效果较优,对目标化合物进行分离和纯化,选取GCB、PSA、C18和MgSO4等填料为分散萃取(dSPE)材料,采用正交试验设计研究方法,通过基质效应和回收率评价净化效果。
选取茶叶胺菊酯阴性样品,根据上述1.5样品前处理制备茶叶空白基质,以茶叶空白基质为溶剂配置成系列浓度的基质加标溶液,以胺菊酯与TPP内标物的峰面积比值作为纵坐标,胺菊酯浓度与TPP内标物浓度的比值为横坐标,绘制基质匹配标准曲线和溶剂标准曲线。基质效应以η表示:η=(基质匹配标准曲线的斜率k(s+ck)-溶剂标准曲线的斜率k(s)/溶剂标准曲线的斜率k(s)[9]。
正交设计试验见表3,正交设计试验数据见表4,正交设计试验数据分析见表5。
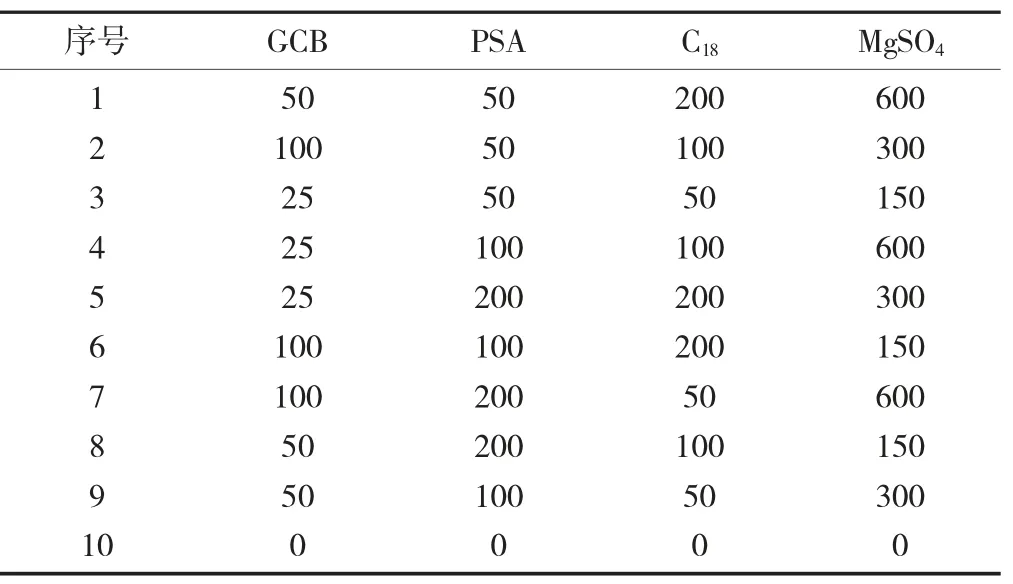
表3 正交设计试验/mg

表5 正交设计试验数据分析/mg
由表4可知,GCB,PSA,C18和MgSO4等净化材料对胺菊酯的基质效应显著,为进一步明确GCB,PSA,C18和MgSO4各因素中对基质效应的影响,采用直观分析法对表4试验结果进行处理。正交试验数据处理过程如下:分别对每次试验各因素的一水平的试验结果求和,即K1,再对每次试验各因素的二水平的试验结果求和,即K2,对每次试验各因素的三水平的试验结果求和,即K3,分别求出各因素的极差R(最大值和最小值之间的差值),根据极差R的大小判断各因素对试验结果的影响。

表4 正交设计试验数据
由表5可知,R(GCB)>R(PSA)>R(C18)>R(MgSO4),极差越大,所对应的因素越主要,由此可以确定GCB和PSA在净化过程中为主要因素,C18和MgSO4在净化过程中为次要因素,为寻找最优的水平组合,首先对GCB进行单因素试验,再对PSA进行单因素试验。
由表5可知,K1(GCB)>K2(GCB)>K3(GCB),由此可见,GCB含量越低,基质效应越显著,为了降低基质效应,试验提高GCB的含量。
GCB单因素试验设计GCB单因素试验设计见表6,GCB对胺菊酯基质效应的影响见表7。

表6 GCB单因素试验设计/mg
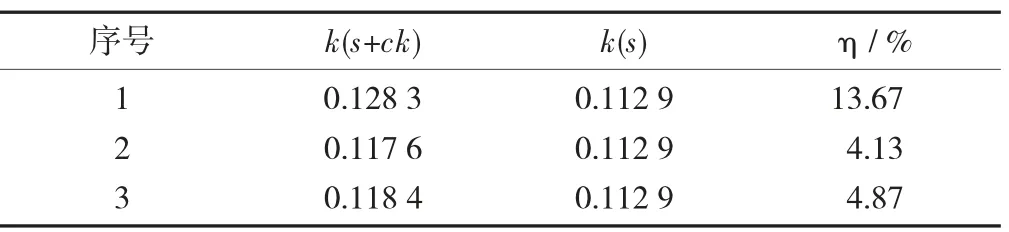
表7 GCB对胺菊酯基质效应的影响
由表7可知,GCB含量增加,基质效应减少,当GCB含量继续增加,大于300 mg时,基质效应无显著变化,由此GCB选定为200 mg。
由表5可知,K1(PSA)=K2(PSA)>K3(PSA),由此可见,PSA含量增加,基质效应下降,为了降低基质效应,试验提高PSA含量。
PSA单因素试验设计见表8,PSA对胺菊酯基质效应的影响见表9。

表8 PSA单因素试验设计/mg
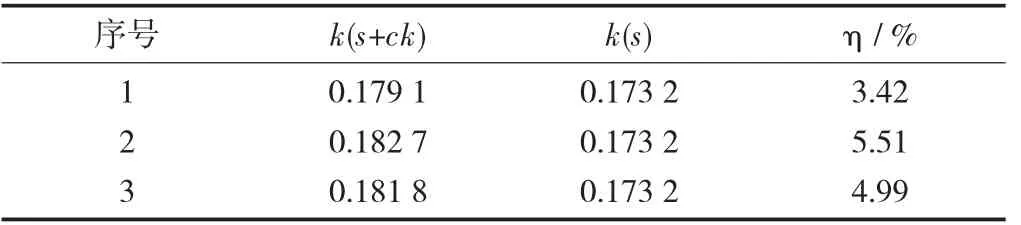
表9 PSA对胺菊酯基质效应的影响
由表9可知,当PSA>300 mg,含量继续增加,基质效应无显著变化,由此PSA选定为300 mg。
结合表4、表5、表7和表9数据结果和数据分析,分散萃取材料的配比为GCB(200 mg),PSA(300 mg),C18(200 mg),MgSO4(300 mg)。
2.3 方法验证与评价
2.3.1 方法线性方程、检出限和定量限
采用茶叶空白基质配制标准溶液,以胺菊酯与内标物质的质谱响应峰面积比值(Ri)作为纵坐标,质量浓度作为横坐标绘制标准工作曲线(由于样品和标准溶液的内标物质量浓度一致,采用质量浓度为横坐标),标准工作曲线为Y=0.003 630 96X+0.001 422 55,在2~100 μg/L的质量浓度范围内呈现良好的线性关系,相关系数R2=0.994 2。胺菊酯采用茶叶空白基质逐级稀释,当质量浓度稀释至3.3 μg/L,S/N的值为3,由此确定方法检出限(LOD)为3.3 μg/kg,以S/N=10计算最低添加水平(10 μg/kg),以10 μg/kg为添加水平进行回收率试验,根据最低添加水平的回收率和相对标准偏差确定方法定量限(LOQ)为10 μg/kg。
2.3.2 方法准确度和精密度
分别称取2.0 g胺菊酯茶叶阴性样品,加入适量标准使用溶液,配制成低、中、高3个质量浓度水平(10.0,20.0,100.0 μg/kg)的加标样品,按照1.5样品前处理方法进行提取与净化,然后采用UPLC-MS/MS检测,每一个加标水平测定6次。
胺菊酯在茶叶中的加标回收率(n=6)见表10。

表10 胺菊酯在茶叶中的加标回收率(n=6)
由表10可知,胺菊酯在低、中、高3个质量浓度水平均有着较好的准确度与精密度,加标回收率为77.11%~105.71%,相对标准偏差(RSD)为6.39%~7.84%。
2.4 实际样品检测
采用QuEChERS-超高效液相色谱-串联质谱法对30份绿茶、红茶、青茶、黑茶和白茶样品进行监测,其中胺菊酯阳性样品1份,残留量为0.012 mg/kg,检出率为3.33%,茶叶中胺菊酯的检出率和残留量均较低。
3 结论
以乙腈(1%醋酸)为提取溶剂,振荡为提取方式,经石墨化碳黑、PSA和C18净化,通过前处理方法中净化材料的优化,选择最优的净化材料配比,建立QuEChERS-超高效液相色谱-串联质谱法检测茶叶中胺菊酯的试验方法。该分析方法高效便捷、可操作性强,方法的灵敏度、精密度和准确度满足茶叶的日常检测要求。
