Increased monoamine oxidase activity and imidazoline binding sites in insulin-resistant adipocytes from obese Zucker rats
2022-02-15ChristianCarpLucMartiNathalieMorin
Christian Carpéné, Luc Marti, Nathalie Morin
Christian Carpéné, Luc Marti, Nathalie Morin, Institut des Maladies Métaboliques et Cardiovasculaires, INSERM, Toulouse 31342, France
Nathalie Morin, Faculté de Pharmacie de Paris, Paris University, Paris 75270, France
Abstract BACKGROUND Despite overt insulin resistance, adipocytes of genetically obese Zucker rats accumulate the excess of calorie intake in the form of lipids.AIM To investigate whether factors can replace or reinforce insulin lipogenic action by exploring glucose uptake activation by hydrogen peroxide, since it is produced by monoamine oxidase (MAO) and semicarbazide-sensitive amine oxidase (SSAO) in adipocytes.METHODS 3H-2-deoxyglucose uptake (2-DG) was determined in adipocytes from obese and lean rats in response to insulin or MAO and SSAO substrates such as tyramine and benzylamine. 14C-tyramine oxidation and binding of imidazolinic radioligands [3H-Idazoxan, 3H-(2-benzofuranyl)-2-imidazoline] were studied in adipocytes, the liver, and muscle. The influence of in vivo administration of tyramine +vanadium on glucose handling was assessed in lean and obese rats.RESULTS 2-DG uptake and lipogenesis stimulation by insulin were dampened in adipocytes from obese rats, when compared to their lean littermates. Tyramine and benzylamine activation of hexose uptake was vanadate-dependent and was also limited, while MAO was increased and SSAO decreased. These changes were adipocyte-specific and accompanied by a greater number of imidazoline I2 binding sites in the obese rat, when compared to the lean. In vitro, tyramine precluded the binding to I2 sites, while in vivo, its administration together with vanadium lowered fasting plasma levels of glucose and triacylglycerols in obese rats.CONCLUSION The adipocytes from obese Zucker rats exhibit increased MAO activity and imidazoline binding site number. However, probably as a consequence of SSAO down-regulation, the glucose transport stimulation by tyramine is decreased as much as that of insulin in these insulin-resistant adipocytes. The adipocyte amine oxidases deserve more studies with respect to their putative contribution to the management of glucose and lipid handling.
Key Words: Obesity; Adipocyte; Amine oxidases; Imidazoline binding sites; Creatine kinase B; Idazoxan; Lipogenesis; Hydrogen peroxide; Glucose uptake
INTRODUCTION
Alongside the numerous models of transgenic mice exhibiting an overt obese phenotype, the Zucker fatty rat remains since decades one of the most widely studied animal models of genetic obesity. Obesity in these rats is inherited as an autosomal recessive trait, as initially reported in 1961 by Zucker and Zucker[1]. It was in 1996 that independent studies demonstrated the molecular basis of such obesity, which does not need any challenge with hypercaloric diet to develop spontaneously[2-4]. Affected rats have a missense mutation in the leptin receptor and show hyperphagia together with various endocrine and metabolic alterations similar to those that appear in the human metabolic syndrome. Leptin is an adipokine produced by adipose tissue and it plays an important role in the central regulation of energy balance[5], together with other functions (e.g., in reproduction). Leptin activates its own receptors in the brain, thereby decreasing energy intake and increasing energy expenditure. Thus, the obese Zucker rats (previously called recessive homozygous fa/fa fatty rats) spontaneously exhibit bulimia and obesity and rapidly become hyperinsulinemic. Consequently, they have been considered as a useful model of insulin resistance and of dyslipidemia with related kidney diseases[6]. Nevertheless, the usefulness of Zucker fatty rats as a model of type 2 diabetes is questionable, since these hyperphagic animals have only mild glucose intolerance, at the expense of dramatically increased insulin release by endocrine pancreas. Indeed, it has been reported that, during the early onset of obesity, the Zucker fatty rats are hypersensitive to insulin[7] and more capable to store lipids than their lean littermates[8]. One assumes that, once adult and obese, these rats develop alternative pathways allowing a still efficient storage of ingested energy in the form of triacylglycerols in the large lipid droplets of their adipocytes[9]. The impressive adipogenic and lipogenic activities maintained throughout the growth of fatty rats, boosted by an excessive energy intake, explains how 40% of body weight is composed of lipids when the Zucker rats reach 14 wk of age[9]. As in obese patients,the adipose depots of mature obese rats are hypertrophied despite established insulin resistance, which results in a lowered sensitivity of the adipocytes to anabolic actions of insulin such as lipogenesis stimulation[10]. The obese Zucker rat is, by definition,also resistant to leptin, and the obese animals present higher circulating levels of this adipokine compared to their lean littermate[11]. Our primary objective was to detect what type of adaptation might occur in adipocytes to circumvent such insulin/leptin resistance in order to maintain a still efficient fat deposition.
It has been reported that pharmacological adrenergic intervention on obese Zucker rats, such as treatment with the β-adrenergic agents celiprolol or clenbuterol[12,13],mitigates their insulin resistance, probably by improving insulin-stimulated glucose uptake in skeletal muscles and adipose tissues. The improvements were not accompanied with a notable body weight loss but were related to a tendency to normalize the repartition of energy fluxes between fat and muscular tissues[14]. To support such beneficial effect of reinforcing adrenergic inputs in the obese Zucker rat,was the demonstration that, in the adipose tissue of young suckling littermates, there was already, at 2 wk of age, a difference in the β-adrenergic signaling pathway in white adipocytes between the future obese and the future lean[15]. It is therefore admitted that impairment of the responsiveness to catecholamines plays a role in worsening the obesity syndrome triggered by leptinergic system invalidation. This led us to further study the alterations of biogenic amine pathways in obese Zucker rats,since these metabolites have a potential role as biomarkers of the metabolic syndrome[16].
It has been already reported that the basal lipolytic activity is higher in adipocytes from obese rats, and that resistance to the lipolytic action of catecholamines appears to counteract such elevated baseline lipolysis[17]. This decrease of β-adrenergic responsiveness seems therefore to be an adaptive mechanism aiming at moderatingin vivoincrease in plasma free fatty acids (FFA). In fact, any excessive increase in circulating FFA is deleterious as it contributes to the complications of insulin-resistant states, such as hepatic steatosis. In addition to their important role in lipid mobilization, the catecholamines also activate glucose utilization in muscles[18] and in brown adipose tissue (BAT)[19]. In a recent study, we observed that high doses of catecholamines mimic the stimulatory effects of insulin on glucose transport, at leastin vitroin rodent isolated adipocytes[20]. All these observations indicated that amines might constitute factors other than insulin capable of affecting directly glucose utilization in fat cells.More precisely, the insulin-like effect of high doses of catecholamines was independent from adrenoceptor stimulation and was rather depending on the presence of the tyrosine phosphatase inhibitor vanadium[20]. In this complex situation, we focused our comparison between lean and obese Zucker rats on the still elusive influence of amines on adipose cell biology.
Alongside their activation of receptors on pre- and post-synaptic cell types,catecholamines are exquisitely regulating various adipocyte functions,viatheir rapid release from presynaptic vesicles or other supposed stores[21,22] and their prompt turn-off mechanisms, involving reuptake, degradation, and desensitization to avoid overstimulation. One of the major catabolic steps of catecholamine degradation is catalyzed by monoamine oxidase (MAO). When oxidating any given substrate, MAO does not only terminate its neurotransmitter function, it also releases the corresponding aldehyde and hydrogen peroxide. The MAO activity has been reported to be lower in the liver of the obese than in the lean rat[23], but to our knowledge, no report has described so far a change of this mitochondrial enzyme in adipose depots.
The following results will compare in obese and lean Zucker littermates, the metabolic effects of a MAO substrate, tyramine, as well as its oxidation by MAO and another amine oxidase highly expressed in fat cells, the semicarbazide-sensitive amine oxidase (SSAO). The data presented thereafter will also indicate that the imidazoline binding sites (I2-sites) that have been documented to be present on MAO[24-27] are more numerous in the white adipose tissue (WAT) of the obese than in the lean rat.
MATERIALS AND METHODS
Chemicals
2-1, 2-3H-deoxy-D-glucose (2-DG; 10 Ci/mmol), 3-3H-D-glucose (10 Ci/mmol), and14C-tyramine were from PerkinElmer (Shelton, United States).3H-2-(2-benzofuranyl)-2-imidazoline (3H-BFI; 50 Ci/mmol) and3H-Idazoxan (42 Ci/mmol) were purchased from Amersham Bioscience/GE Healthcare in 2004 (Buckinghamshire, England), then isotopically diluted in ethanol and stored at -12 °C, and yearly verified since by high performance liquid chromatography (> 90% pure) before being used as radioligands for binding experiments until 2010. Electrophoresis products and devices were from Bio-Rad (Ivry/Seine, France). Rabbit polyclonal antibodies against MAO and SSAO were kindly given by Pr. A. Parini (Toulouse, France), and by Pr. M. Unzeta(Barcelona, Spain), respectively. Rauwolscine was kindly given by Dr. A. Remaury(Sanofi, Chilly-Mazarin, France). All remaining chemicals and drugs, such as cirazoline, sodium orthovanadate, and bovine insulin, were purchased from Merck(Darmstadt, Germany) or its affiliate Sigma-Aldrich (Paris, France).
Animals
Three sets of experiments were successively performed to explore the imidazoline binding sites and the MAO/SSAO activities in various tissues from Zucker rats, and the functional response of adipocytes. All the comparisons between lean and obese Zucker rats were made between littermates. Such comparative approach between littermates was performed on a number of male and female rats that was not always the same for each genotype in each successive experiment. This was due to the fact that the number of pups of a given litter is not always distributed equally according to the gender and to the obese phenotype (which were stated after weaning only).Consequently, the number and gender of analyzed animals are given thereafter for each subset of experiments. All the rats (Charles River Laboratories, L’Arbresle,France) were housed in hanging wire cages with free access to food and water on a 12 h light/dark cycle (lights on 6 a.m.) at a temperature of 20 °C. They were euthanized after overnight fasting between 9 and 10 wk of age in accordance with the ARRIVE guidelines (Animal Research: Reporting ofIn VivoExperiments)[28].
Adipocyte glucose uptake and lipogenic activity
WAT was removed from intra-abdominal (perigonadic, retroperitoneal, and perirenal;thereafter named visceral WAT) and from subcutaneous (inguinal) locations. It was immediately digested by collagenase in Krebs-Ringer buffer containing 15 mmol/L bicarbonate, 10 mmol/L HEPES, 2 mmol/L pyruvate, and 3.5% bovine serum albumin. Separation, washing, and dilution of the buoyant adipocytes were performed in the immediate continuity at 37 °C with fresh buffer prior to the functional assays,which were performed as previously described[20].
The radiolabeled non-metabolizable analog of glucose,3H-2-DG, was used at 0.1 mmol/L for hexose uptake assays as described previously[20], save that the assays lasted 5 min instead of 10 min for human fat cells, which are less metabolically active.
Lipogenic activity was determined by measuring the radioactivity incorporated into cellular lipids after 120-min incubation with the indicated agents and 0.5 mmol/L3Hglucose. This very simple bioassay, primarily designed by Moody and coworkers[29],was performed with slight adaptations[30] in the same plastic vial, which was used for incubation, extraction, and scintillation counting, since the3H-glucose that was not metabolized by the fat cells remained in the lower phase and could not excite the nonwater-miscible liquid scintillation cocktail (InstaFluor-Plus, PerkinElmer, Waltham,United States) of the upper phase, containing the neosynthesized lipids, as already demonstrated[31].
Immunoblots
Homogenates were prepared in RIPA buffer, and proteins were solubilized in loading buffer (60 mmol/L Tris-HCl with 2% SDS, 10% glycerol, 1% β-mercaptoethanol, pH 6.8) at 100 °C for 5 min prior to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. After migration, proteins were transferred to polyvinylidene difluoride membranes with a semidry electroblotter (Trans-blot, Bio-Rad). The blots were blocked in wash buffer (50 mmol/L Tris, 200 mmol/L NaCl, 0.1% Tween 20, pH 7.5)with 5% nonfat dried milk for 1 h at room temperature. Then they were incubated overnight at 4 °C with rabbit polyclonal antisera (1:1000) obtained from rabbits immunized against an epitope of MAO-A and MAO-B as described previously[32].After washing, the blots were incubated with peroxidase-labeled anti-rabbit IgG(1:7000) in wash buffer for 60 min. The immunoreactive proteins were detected using enhanced chemiluminescence (Pharmacia Biotech, Piscataway, NJ). Similar procedure was followed when using an anti-SSAO polyclonal antibody developed and tested previously[33]. β-actin amount was determined in stripped membranes, and as no change was observed between preparations from lean and obese animals (not shown),no normalization to β-actin was performed.
Amine oxidase assay
Amine oxidase activity wasmeasured on homogenates of thawed tissues with14Ctyramine in 200 mM phosphate buffer in the presence of an antiprotease cocktail from Sigma as previously described[30]. MAO activity was defined as oxidation inhibited by pargyline 0.1 mmol/L, while SSAO activity was sensitive to semicarbazide 1 mmol/L. BAT was removed from the core of the interscapular fat depot, and the soleus muscle was chosen as representative of skeletal muscles. Like liver samples,these tissues were snap-frozen without buffer and homogenates were prepared on thawed samples as already described[34]. Protein content was measured with DCprotein assay from Bio-Rad. In the case of amine oxidation by intact fat cells, 1 mM of an isotopic dilution of14C-tyramine was incubated for 30 min with adipocyte suspension (~10 mg of cell lipids) in 400 µl of KRBH medium instead of phosphate buffer, and treated as above stated for homogenates.
Radioligand binding
For saturation-binding isotherms, membranes were incubated with increasing concentrations of3H-idazoxan at room temperature for 45 min in Tris-HCl buffer, pH 7.4. The presence of 10 µmol/L rauwolscine was used to mask α2-adrenoceptors as already demonstrated[32]. For3H-BFI, it was not necessary to preclude binding to adrenergic receptors since this imidazolinic radioligand has been shown to be selective for the I2-sites[30,35,36]. In both cases, incubation was stopped by vacuum filtration through Whatman GF/C microfilters with extensive washes with the same buffer at 4 °C.Nonspecific binding was defined in the presence of 100 µmol/L cirazoline and represented 30% to 50% of the total binding depending on the radioligand nature, its concentration, and the biological material tested. Radioactivity bound on the filters was counted in a liquid scintillation spectrometer (Packard, model Tri-Carb 4000) with 4 mL of Packard Emulsifier-Safe scintillation fluid per scintillation microvial.
Statistical analysis
Results are presented as the mean ± SEM of (n) observations. Statistical analyses for comparisons between parameters were performed using ANOVA followed by posthoc Dunnett's multiple comparisons test, and were performed with Prism 6 for Mac OS X (from GraphPad software, San Diego, CA).
RESULTS
Hyperinsulinemia and insulin resistance of adipocytes are characteristic traits of the obese Zucker rat
Some characteristic features of the obese Zucker rats were first verified. At the age of 9-10 wk, the obese rats exhibited larger body weight and adiposity, with elevated insulin and triacylglycerol plasma levels, when compared to their lean littermates of the same age (Table 1). Fasting plasma glucose levels were slightly higher in the obese group, but with a less pronounced difference with respect to the other circulating parameters influenced by genotype. Blood glucose was 1.12 times higher in obese rats,while insulin was four to five times higher and triacylglycerol two times higher than in lean controls. As expected, the males bearing the two alleles with the mutated leptin receptor (fa/fa) were hyperphagic since their daily food consumption was increased by 1.3 times,i.e., similarly to their increased body weight, while the sum of their dissected fat depots was five-to-six-fold heavier than in lean littermates (Table 1).

Table 1 Biological parameters of the lean and obese rats used in this study
Our descriptive approach readily confirmed in white adipocytes the insulinresistant state of the obese rats, which accompanies their clear-cut hyperinsulinemia.The hexose uptake assays showed that the maximal effect of insulin (obtained at 0.1µmol/L-1 µmol/L) was one-half lower in obese than in lean rats (Figure 1A). The white adipocytes of obese rats also exhibited a reduced sensitivity to insulin: Their response to 10 nmol/L insulin represented less than 60% of the maximal effect of the pancreatic hormone in obese rats while it reached 80% in lean controls (Figure 1A). Asimilar pattern was obtained when lipogenic activity of the adipocytes was assessed by the incorporation of radioactivity into cell lipids after 120-min incubation with 3-3H-Dglucose (Figure 1B). The insulin resistance was not totally complete in these young obese rats, since the baseline incorporation was increased by a six-fold factor in response to 100 nmol/L insulin. Nevertheless, this maximal activation reached a 12-fold factor in adipocytes from lean rats (Figure 1B). As for hexose transport, the lipogenic effect of 10 nmol/L insulin reached 42% ± 8% and 86% ± 7% of maximal response in obese and lean rats, respectively (n= 8 and 6, respectively,P< 0.002).

Figure 1 Influence of obesity on the dose-dependent responses of rat adipocytes to insulin activation of hexose uptake and of lipogenesis.

Figure 2 Insulin-like effects of vanadium combined with hydrogen peroxide or with amine oxidase substrates on hexose uptake into rat adipocytes.
In order to bring evidence that other factors other than insulin may help the adipocytes in accumulating large amounts of lipids during the growth of the insulinresistant obese rat, vanadium was tested alone and with hydrogen peroxide, either when directly added at 1 mmol/L, or when generated endogenously by the adipocytesviathe oxidation of the MAO and SSAO substrates tyramine and benzylamine at 1 mmol/L (Figure 2). Sodium orthovanadate was inefficient at 100 µmol/L, either on basal or on insulin-stimulated 2-DG uptake. However, vanadium potentiated the insulin-mimicking action of 1 mmol/L hydrogen peroxide. The combination of these two agents is known to generate peroxovanadate, a powerful protein tyrosine phosphatase inhibitor[37,38]. It led to a stimulation of glucose uptake equivalent to 70%-80% of the response to insulin in lean rats (Figure 2). Tyramine, a substrate of both MAO and SSAO in rodents[34], was also able, when combined with vanadium, to reproduce 60%-70% of insulin maximal response in the lean rats. This insulin mimicry was also obtained with 1 mmol/L benzylamine, but it represented 84% ± 16% of insulin + vanadate effect in lean and only 38% ± 7% in obese rats (n= 5,P< 0.03). As expected, the MAO and SSAO inhibitor phenelzine abolished the effects of tyramine or benzylamine plus vanadium in both genotypes (Figure 2).
These first observations indicated that the MAO- or SSAO-mediated effects of tyramine on glucose handling by fat cells were limited similarly as those of insulin in obese Zucker rats. However, since the effect of benzylamine plus vanadate seemed to decrease even more with obesity, we further studied the protein expression of the amine oxidases in WAT.
Western blot analysis of amine oxidases in adipose tissues of lean and obese Zucker rats
When the expression of MAO and SSAO was determined by Western blot in subcutaneous WAT, opposite changes were observed for these membrane proteins.WAT expressed less SSAO in obese than in lean rats, at least in the subcutaneous depots of 10-wk-old animals (Figure 3). This lower abundancy was agreeing with the above reported lower insulin-like effects of benzyamine regarding glucose uptake in adipocytes. More puzzling was the obesity-related increase in MAO-A protein, while MAO-B was poorly affected in obese rats. As a consequence of these opposite regulations, the resulting effect on glucose transport of the MAO and SSAO substrate tyramine did not exhibit a dramatic change in adipocytes from obese rats, at least when normalized to the maximal insulin response. To state whether tyramine catabolism is really greater in WAT from obese rats, it was decided to investigate its oxidation in various tissues.
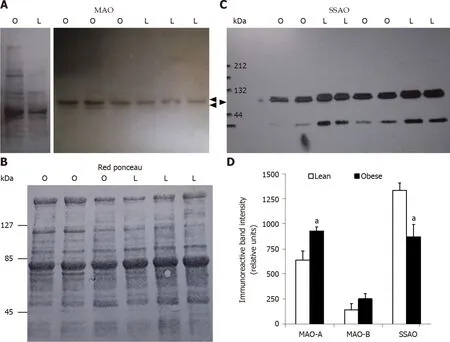
Figure 3 Increased monoamine oxidase and decreased semicarbazide-sensitive amine oxidase protein expression in subcutaneous adipose tissue from obese rats when compared to lean littermates.
Amine oxidase activities in several tissues of lean and obese Zucker rats
The oxidation of 0.5 mmol/L14C-tyramine by crude membranes of different tissues is shown in Figure 4, in which 'total oxidation' means spontaneous oxidation of the amine in the presence of biological material, without any added inhibitor or catalyst.Since the liver is known for being rich in MAO activity, it was tested here as a positive control. As expected, the liver was the richest of the tested tissues regarding MAO activity, which entirely supported total tyramine oxidation, and which did not exhibit difference between genotypes. No SSAO activity was detectable in liver homogenates regardless of the genotype.

Figure 4 Oxidation of tyramine by membrane preparations from different tissues of obese and lean Zucker rats.
Tyramine oxidation in crude membranes of adipose cells from visceral fat depots was greater in obese than in lean rats (Figure 4). This was largely due to an increased MAO activity, which was prominent in white adipocytes when considering the large proportion of total tyramine oxidation that was inhibited by pargyline (therefore MAO-dependent). The smaller fraction of tyramine oxidation that was sensitive to semicarbazide (therefore SSAO-dependent) was not clearly reduced in visceral adipocytes from obese rats (Figure 4). Altogether, these changes were similar to those observed in immunoblots of subcutaneous WAT.
In the interscapular adipose tissue, which contains brown adipocytes, thereby considered as BAT, increased tyramine oxidation was also found in obese rats(Figure 4). Although both MAO and SSAO activities were higher in BAT from obese than from lean rats, none of them reached the levels found in WAT. Lastly, in skeletal muscle crude membranes, tyramine oxidation was weak regardless of the genotype.
The fact that an increase in tyramine oxidation only occurred in WAT and BAT raised a concern about possible yield variations during membrane preparation between obese and lean rats. In fact, only the adipose depots were impressively much fatter in the obese than in the lean rat, while this was less evident for the liver and skeletal muscles. To circumvent a possible bias generated by an excess of fat in the different centrifugation steps necessary for membrane preparation, we studied the genotype influence on the capacity to oxidize tyramine in undamaged, functional adipocytes freshly isolated from visceral WAT. Again, adipocytes from obese rats possessed a higher capacity to oxidize tyramine when compared to lean ones(Figure 5). Even being of larger cell size, the adipocytes of obese Zucker rats definitely exhibited a larger MAO activity than lean controls. Figure 5 also shows that the same SSAO activity was found in adipocytes from obese rats and lean controls. Finally,MAO and SSAO activities were complementary to account for the total tyramine oxidation performed by adipocytes, ruling out a notable contribution of other amine oxidases.

Figure 5 Tyramine oxidation in adipocytes from obese and lean Zucker rats.
In view of the enlarged MAO activity that appeared specific for WAT and BAT, we reanalyzed in various tissues from obese and lean rats an unpublished comparison of the population of I2imidazoline binding sites, previously described to be present on MAO enzymes[24,26,27] and distinct from the I1sites, essentially labeled by clonidine or moxonidine[39,40].
Pharmacological analysis of imidazoline binding sites in tissues of lean and obese Zucker rats
By using a saturating concentration of3H-BFI (20 nmol/L), we quantified the I2imidazoline binding sites in crude membranes from the same anatomical locations as above. A difference in the amount of3H-BFI bound was evident between obese and lean rats in visceral adipocytes only. The obese rats exhibited the same3H-BFI binding as the lean controls in the liver, interscapular BAT, and skeletal muscle (Figure 6).
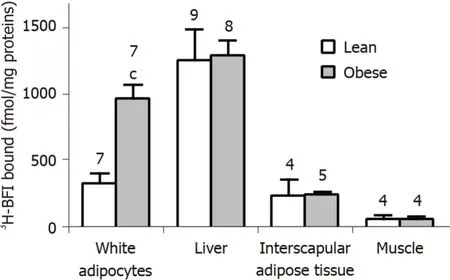
Figure 6 Tissue-selective larger imidazoline binding site density in the white adipocytes from obese rats when compared to lean littermates.
This comparison was completed by saturation binding experiments in adipocyte membranes from visceral and subcutaneous WAT with3H-BFI and3H-idazoxan.Increasing concentrations of3H-BFI (from 0.2 to 26 nmol/L) resulted in saturation curves for total binding, with a clearly lower non-specific binding that was defined in the presence of 100 µmol/L cirazoline (Figure 7). Scatchard plots of the resulting specific binding gave an almost linear relationship between the bound/free ratio and the quantity of bound radioligand (Figure 7). The estimates of the affinity constant (KD)and of the population density (Bmax) of the corresponding single class of binding sites are reported in Table 2. A significant increase of the number of I2imidazoline binding sites was found in WAT of the obese Zucker rats when compared to lean controls. The nature of these binding sites was not modified by the obesity status since the KDvalues were not different between obese and lean rats (Figure 7 and Table 2).

Figure 7 Saturation binding analysis with 3H-2-(2-benzofuranyl)-2-imidazoline and 3H-idazoxan in adipocyte membranes from white subcutaneous adipose tissue of lean and obese rats.
Similar saturation binding parameters were observed when using3H-idazoxan(from 0.7 to 45 nmol/L) as radioligand in the presence of 10 µmol/L rauwolscine to preclude its binding to α2-adrenoceptors. Again, higher Bmaxvalues were found in the WAT of obese rats without notable change in KDvalues when compared to lean controls (Figure 7 and Table 2).3H-idazoxan exhibited a lower affinity for the I2sites than3H-BFI, and it exhibited also a less selectivity towards imidazoline sites since, at 14 nmol/L, non-specific binding reached 50% of total binding in lean while it represented 10% of total binding in obese rats (not shown).
Nevertheless, the Bmaxvalues obtained with3H-idazoxan were of the same order of magnitude as the values observed with3H-BFI, suggesting that both imidazolinic radioligands recognized the same binding sites. Moreover, the observed increase in the density of I2sites in obese rat could not be associated to changes in the protein amounts used in binding experiments since we used similar protein levels regardless of the genotype, at least for visceral WAT (Table 2).
Thus, the larger density of I2sites specifically found in the WAT of obese rats was related to an increased MAO activity. Binding data were also in agreement with the previously reported capacity of BFI to inhibit MAO activity present in human and rodent adipose tissues[30]. To further explore the functional link between I2sites,MAO activity, and tyramine insulin-like effect, we performed two distinct approaches,one consisting in a short-termin vitroexperiment on the interplay between I2binding sites and MAO activity, and the other based on repeatedin vivoadministration of tyramine plus vanadate to Zucker rats.
Since MAO activity and I2binding sites appeared to be linked with respect to their higher levels in WAT from obese rats when compared to lean controls, we took advantage of their abundancy in the liver to verify whether the one can influence the other. Figure 8 shows the total and the non-specific binding of increasing doses of3Hidazoxan to liver crude membranes from lean Zucker rats. The former followed a curve reaching saturation while the latter was linearly increasing with radioligand concentration. Addition of a large dose of tyramine (5 mmol/L) to saturate the active sites of hepatic MAO abolished the total idazoxan binding without altering the nonspecific one. This confirmed the functional interaction between MAO activity and I2binding sites.
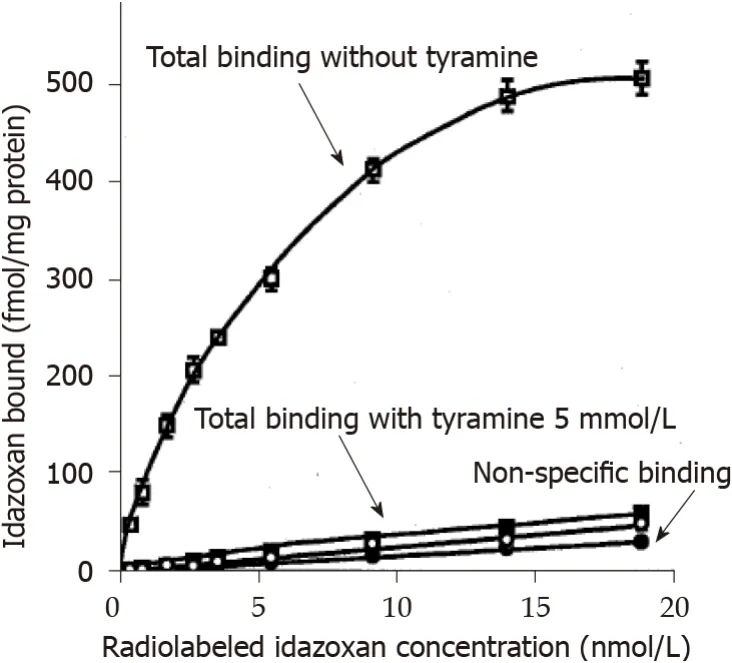
Figure 8 Influence of tyramine on idazoxan binding to liver crude membranes.
In vivo treatment of lean and obese rats with tyramine plus vanadate
A repeated treatment with 'tyramine + vanadate' was performed on 9-wk-old lean and obese rats to investigate whether thein vitroinsulin-mimicking effects of this combination could have anyin vivorelevance. The subcutaneous administration of tyramine at 3 mg/kg/d combined with sodium orthovanadate at 0.3 mg/kg/d for 1 wk was well tolerated, and did not modify significantly the body weight gain in obese rats when compared to controls receiving the vehicle (daily s.c. bolus of 0.3 mL NaCl 0.9%): 47 + 8vs46 ± 8 g (n= 3-4, NS). Body weight gain was also unaltered in treated lean rats (33 ± 3vs32 ± 3 g;n= 3-4, NS). Daily food intake was also unchanged since it remained around 23 g/rat in the treated and control lean groups, and was comprised between 29 and 40 g/rat in the obese groups (not shown). Obviously, the clear-cut difference between obese and lean rats regarding fat deposition could not be modified by this relatively short-term treatment. Only a non-significant tendency to enhance the mass of dissected adipose depots was seen in obese treated rats, while one of the dissected fat depots was heavier in lean rats receiving tyramine (Figure 9). Interestingly, tyramine + vanadate treatment reduced moderately but significantly the fasting plasma levels of glucose and triacylglycerols in the obese treated group (Figure 9).Regarding glucose uptake, the tyramine + vanadate treatment was not sufficient to mitigate the clear insulin-resistant state of the adipocytes of obese Zucker rats(Figure 9). However, it was investigated whether the insulin-effect of peroxovanadate on glucose transport in adipocytes (generated byin vitroaddition of 1 mmol/L hydrogen peroxide + 0.1 mmol/L vanadate) was modified by the tyramine + vanadate treatment, especially in the obese rats, in view of their higher MAO content. The hexose uptake stimulated by hydrogen peroxide plus vanadate was similar in control and treated obese rats: 4.19 ± 1.30 and 3.29 ± 1.55 nmol 2-DG/100 mg lipids/5 min,respectively (n= 5, NS). Again, this peroxovanadate-dependent stimulation of hexose uptake was higher in lean rats, regardless of treatment: 11.32 ± 1.26 and 10.46 ± 2.61 nmol 2-DG/100 mg lipids/5 min (n= 5, NS). Nevertheless, it is worth noting that these values were, in each genotype, close to the maximal stimulation of hexose uptake triggered by high doses of insulin (see Figure 9). At least, the tyramine + vanadate treatment did not induce any desensitization of the insulin mimicking action of hydrogen peroxide plus vanadate.

Figure 9 Influence of ‘tyramine + vanadate’ treatment on adiposity, glucose and lipid handling, and insulin responsiveness of adipocytes in lean and obese rats.
Thus, the effects ofin vivotyramine + vanadate treatment could not reveal relevant antidiabetic or anti-obesity properties, at least at the dose and duration tested.However, the limited - but encouraging - beneficial influence of this treatment on hyperglycemia and hypertriglyceridemia leaves opens the possibility to improve metabolic control by providing MAO substrates, which might enhance glucose utilization in fat depots and facilitate glucose handling at the expense of a slightly larger fattening.
DISCUSSION
The first unexpected observation of this comparison between obese and lean Zucker rats was that the stimulatory effect of millimolar dose of tyramine + vanadate on glucose uptake in adipocytes was altered as much as that of insulin in the obese rat.
Obviously, insulin resistance of adipocytes from obese Zucker rat was expected, and fully confirmed here, but it could be supposed that agents that act independently from insulin receptor activation were less hampered with obesity.
Indeed, many factors could be involved in the decreased insulin-like effect of the combination tyramine + vanadate or benzylamine + vanadate on adipocytes from obese rats, such as altered glucose transporter equipment and recruitment[41].Anyhow, our initial working hypothesis was based on a putative reduction in the amount of adipose amine oxidases since we recently reported that mice bearing genetic invalidations of SSAO are obese[31].
The second major finding was that, contrarily to our assumptions, MAO activity was higher in the WAT of obese rats than in lean ones. In other words, the MAO substrate tyramine was not more efficient in activating glucose utilization while its degradation was increased in adipocytes from obese animals. This could have a sense if tyramine effect was mediated by a receptor-dependent mechanism, thereby considering that its increased degradation hampered receptor activation. But, in the case of tyramine, the insulin-like effect on glucose utilization by rat adipocytes is mediated by amine oxidation and subsequent hydrogen peroxide release[34]. This is also the case for various other amine oxidase substrates, such as benzylamine or methylamine, which are also active in rodent[42] and human adipocytes[43].
Although the insulin-mimicking action of tyramine is tremendously potentiated by vanadium[34], it was amazing to observe a decrease of such MAO-mediated response together with an increase of MAO activity in the adipocytes from obese rats. We confirmed the latter increase with regard to the level of enzyme activity, protein immunoreactivity, and an increased number of I2imidazoline binding sites, located on this mitochondrial enzyme[24,25,32]. Unfortunately, we encountered some technical difficulties with the quantification of rat MAO mRNA despite our know-how developed on MAO expression in the mouse WAT[31]. Consequently, we cannot add to the above converging data that the expression of the genesMaoaandMaobis increased in the WAT of obese Zucker rats. Despite such limitation, our study clearly indicates that the obesity-related increase of MAO activity was selective of WAT and of BAT, since no change was detected in the liver or in skeletal muscle. Moreover,MAO up-regulation was not paralleled by similar changes of SSAO in WAT. This will be discussed below after summarizing other major metabolic and endocrine defects of the obese Zucker rats that have been confirmed in the present study.
The overweight of the obese rats used for this comparative approach, and the stabilization of their fasting blood glucose close to the normal range at the expense of dramatically increased insulin levels are characteristic traits of this obesity model,which were highly similar to those primarily described for over half a century in the Zucker fatty rat[44]. While the original pioneering findings demonstrated the insulin resistance of skeletal muscles from obese Zucker rats, we confirm here the insulin resistance of white adipocytes at the level of glucose transport and lipogenic activities.More importantly, such insulin resistance was associated in WAT to dysregulations not described so far.
Thus, MAO activity, MAO immunoreactive protein amount, and imidazoline I2-binding site population were increased in adipocytes from the obese Zucker rats. In muscles of the same animals, the low expression of MAO and SSAO, already documented by diverse techniques[45,46], was unmodified in spite of similar insulin resistance regarding glucose utilization[44]. Facing to such tissue-specific difference, it was therefore difficult to establish a mere link between MAO expression and insulin sensitivity. Although the mechanisms underlying the overexpression of MAO activity specifically in WAT and BAT could not be deciphered here, it must be reminded that MAO is regulated in various organs by numerous factors, including aging, and in different directions for A and B forms[27]. This complexity of regulations may explain why the decreased hepatic MAO previously found in obese rats[23] was not confirmed here. Other possible explanation of this discrepancy can be that in the previous study,it was the decreased mitochondrial content per mass of fatty liver that was the drive for the observed reduction, while in the present study MAO activity was expressed per mass unit of crude membrane proteins. In WAT and BAT, whether adipose MAO was up-regulated by an excess of its substrates as a consequence of the hyperphagia driving obesity remains speculative, and has no rationale to be different from the contradictory changes found in the liver.
Closely related to the MAO increase found in the adipose tissue of obese rats was the increase in imidazoline I2binding sites. It occurred only in WAT and was readily visualized by a larger Bmaxfor either3H-BFI or3H-idazoxan regardless of fat depot anatomical location. The link between I2sites and MAO was further supported in the liver by two observations. First, a lack of change between lean and obese rats was found for both parameters: Neither MAO Vmaxnor3H-BFI Bmaxwas altered with obesity in the liver. Second, the3H-idazoxan binding to hepatic I2sites was completely prevented by a high dose of tyramine. These observations complete our previous report about the capacity of BFI to inhibit MAO-A and MAO-B activities[30]. The obesity-related changes in imidazoline I2binding sites seemed to be specific of adipose cells, since no change was evidenced in several other peripheral tissues (this study),and in the brain of obese rats when compared to lean controls[47].
Also of interest was that a lower SSAO activity was concomitant to a larger MAO richness in the adipose cells from obese rats. It can be hypothesized that the small reduction in SSAO activity was involved in the resulting weak insulin-like effect of tyramine, since tyramine is a substrate of both enzymes, at least in rats[34]. Thus,according to our working assumptions, SSAO was reduced in WAT from obese rats,and accordingly the activation of glucose uptake by the SSAO substrate benzylamine(plus vanadate) was diminished.
Indeed, reduced expression of SSAO has been already reported at the level of mRNA abundance and of benzylamine oxidation in the WAT of obese rats as compared with their lean littermates[48]. In this study, the SSAO activity found in the WAT of obese rats was one-half lower than that found in lean controls. Although this reduction is apparently greater than that reported here for immunoreactive SSAO,both observations remain in good agreement. Since benzylamine oxidation was measuredviaa fluorometric method quantifying the hydrogen peroxide production in the previous study[48], it can be supposeda posteriorithat this had generated underestimates of SSAO activity in obese rats, in view of the increased catalase activity found in their WAT[49]. Such increased catalase might also impair the hydrogen peroxidedependent activation of glucose uptake that we report here in the obese adipocytes.Whether the decreased SSAO activity in obese WAT is related to a previously reported increase in the spermine and spermidine content of the adipocytes of obese Zucker rats[50] remains to be established, but it must be noted that, alongside their role of regulator of triacylglycerol synthetic enzymes, polyamines are also SSAO substrates.More surely, it can be asserted that the SSAO decrease appeared to be tissue-specific for WAT, as no change in SSAO activity has been found in other anatomical locations(this study), as well as in the aorta of obese Zucker rats[51]. Anyhow, a similar diminution of SSAO has not been found in other animal models of obesity, since modest increases in SSAO activity have been observed in the adipose tissue from db/db mice and dogs fed a high-fat diet when compared to respective control(reviewed in[43]).
It becomes evident that it is not only overweight and WAT hypertrophy that can be related to SSAO expression, but also the overall alteration of glucose and lipid handling of obese states. Of course, SSAO is one of the most abundant proteins present on the cell surface of the adipocytes[52]. SSAO-mediated oxidation of exogenous substrates induces glucose uptake in adipocytes isolated from WAT or obtained byin vitrodifferentiation of preadipose cell lines (reviewed in[43]) and in several other models[53]. SSAO-mediated deamination reactions generate hydrogen peroxide,which also induces antilipolytic and lipogenic effects[54], especially in the presence of vanadate, by forming pervanadate, a potent insulin-like agent[38,55]. More recently, it has been proposed that SSAO interplays with Zinc-α2-glycoprotein[56] and with lipid metabolism in adipocytes[57]. In addition, SSAO is identical to vascular adhesion protein-1 (VAP-1), which supports leukocyte extravasation[58]. These multifunctional facets of SSAO are therefore rendering highly probable the occurrence of various apparently contrasting regulations according to the pathological states of adipose tissues.
Two very recent findings are dealing with our observations of a parallel increase of MAO and I2sites in the WAT of obese Zucker rats.
On the one hand, untargeted metabolomics analysis performed in the blood of Zucker diabetic rats indicates that the tryptophan and tyrosine metabolisms are the most dysregulated pathways in this model[59]. This is somewhat dealing with our description of changes in MAO activity occurring in the insulin-resistant adipocytes from obese rats. First, tryptophan is linked with kynurenine and serotonin, which is readily oxidized by MAO and generates 5-hydroxyindoleacetic acid. Second, tyrosine is the precursor of biosynthetic pathways for catecholamines and trace amines and therefore an important source of MAO substrates. By associating these two independent findings, it can be assumed that both biosynthesis and degradation of catecholamines are altered in the obese Zucker rat. Although poorly described,catecholamine catabolism occurs in adipose tissues, especially in visceral WAT, and has been shown to influence both glucose metabolism[60] and adrenergic contraction of mesenteric arteries[61].
On the other hand, it has been proposed during the completion of this work that creatine kinase B controls futile creatine cycling, and is powerfully induced by thermogenic stimuli in both mouse and human adipocytes[62]. Intriguingly, creatine kinase B has also been proposed to be an imidazoline binding protein in rodents[36].Indeed, "non-MAO" imidazoline binding sites have been evidenced in diverse mammalian tissues, such as those related to brain creatine kinase B[63]. At the present time, one limitation of our study is that we cannot attest that the reported3H-idazoxan and3H-BFI Bmaxvalues are overestimating or not the MAO quantity in the studied peripheral tissues.
Although we have already documented that I2site ligands inhibit MAO[24] and in spite of confirming this link in the present study, it is necessary to remind that we have serendipitously described the antilipolytic properties of 0.1-1 mmol/L BFI in human fat cells without bringing clear demonstration of the underlying mechanisms[30].Whether the "non-MAO" imidazoline binding sites such as creatine kinase B are involved in this effect as well as the obesity-related changes of I2sites in WAT remains to be established.
CONCLUSION
This work is reporting for the first time an adipocyte-specific increase of both MAO and I2sites in obese Zucker rats when compared to their lean controls. In spite of this higher capacity of catabolizing endogenous or exogenous amines, the insulin-like effects of tyramine and benzylamine, decreased as much as the insulin activation of glucose transport in the large fat cells of the obese rat. It remains to establish whether the activation of glucose uptake by MAO and SSAO substrates, which can be readily observedin vitroin the presence of vanadate, really occursin vivoand might help the insulin-resistant fat cells in storing energy under the form of lipids.
ARTICLE HIGHLIGHTS
Research background
The genetically obese Zucker rat is hyperphagic and accumulates the excess of calorie intake in the form of lipids in spite of the insulin-resistant state of its adipocytes.
Research motivation
To investigate what biologi cal events or natural biochemical processes drive the glucose utilization in fat cells of obese Zucker rats, which are not fully responsive to the lipogenic action of insulin.
Research objectives
Hydrogen peroxide is a biological chemical that can mimic several insulin actions on adipocytes, such as stimulating glucose entry and lipogenesis, and inhibiting lipolysis.Since it is a product of various enzymes in adipocytes, we focused our objective in searching whether the expression activity and biological effect of two types of them,namely, the monoamine oxidase (MAO) and semicarbazide sensitive amine oxidase(SSAO), abundant in adipocytes, were modified in obesity states.
Research methods
Experimental methods included Zucker rat husbandry, with obese and lean littermates(the former bearing homozygotous recessive mutation of fa/fa gene), preparations of freshly isolated adipocytes, functional exploration of hexose transport using uptake assays with appropriate pharmacological agents, and determination of lipogenic activity, immunobloting, measurement of amine oxidase activities, and saturation binding analyses.
Research results
There is a good relationship between the increased binding capacity of tritiatedidazoxan and tritiated-(2-benzofuranyl)-2-imidazoline to imidazoline binding sites and the increased MAO-dependent tyramine oxidation in adipose tissue of obese rats.Stimulation of MAO or SSAO by their substrates in the presence of vanadate reproduced approximately two-thirds of the insulin stimulation of glucose uptake in fat cells. However, this insulin-like effect decreased as the insulin responsiveness of adipocytes decreased with obesity.
Research conclusions
It cannot be stated whether the changes in MAO and SSAO expression are a cause or a consequence of the altered glucose handling in the fat cells of obese Zucker rats. At least, the increased tyramine oxidation is found in adipose tissues of the obese rats, not in the liver or in skeletal muscles, and can be associated with the dysregulation of the catecholaminergic system and of the energy balance found in that animal model of genetic obesity.
Research perspectives
The increase of MAO activity in adipose tissue from obese rats has never been reported, at least to our knowledge. Among the limitations of our first description of an increased MAO activity in the adipose tissue of this animal model of obesity, is that we did not decipher the mechanisms supporting such up-regulation, and that it remains unclear whether elevated MAO is a cause or a consequence of the dysmetabolic profile of the Zucker obese rats. This issue deserves further investigations.
ACKNOWLEDGEMENTS
We thank the staff of animal unit CREFRE, headed by Dr. Collet X (Toulouse, France)and Dr. Guerre-Millo M (Paris, France), for their knowledge about Zucker rat physiology. The authors are also indebted for the invaluable technical assistance of Pré vot D & Jousseaume M, and for the hierarchical benevolence of Bouloumié A.
