Current understanding of the role of tyrosine kinase 2 signaling in immune responses
2022-02-15RyutaMuromotoKenjiOritaniTadashiMatsuda
Ryuta Muromoto, Kenji Oritani, Tadashi Matsuda
Ryuta Muromoto, Tadashi Matsuda, Department of Immunology, Graduate School of Pharmaceutical Sciences, Hokkaido University, Sapporo 060-0812, Japan
Kenji Oritani, Department of Hematology, International University of Health and Welfare,Narita 286-8686, Japan
Abstract Immune system is a complex network that clears pathogens, toxic substrates, and cancer cells. Distinguishing self-antigens from non-self-antigens is critical for the immune cell-mediated response against foreign antigens. The innate immune system elicits an early-phase response to various stimuli, whereas the adaptive immune response is tailored to previously encountered antigens. During immune responses, B cells differentiate into antibody-secreting cells, while naïve T cells differentiate into functionally specific effector cells [T helper 1 (Th1), Th2, Th17,and regulatory T cells]. However, enhanced or prolonged immune responses can result in autoimmune disorders, which are characterized by lymphocytemediated immune responses against self-antigens. Signal transduction of cytokines, which regulate the inflammatory cascades, is dependent on the members of the Janus family of protein kinases. Tyrosine kinase 2 (Tyk2) is associated with receptor subunits of immune-related cytokines, such as type I interferon, interleukin (IL)-6, IL-10, IL-12, and IL-23. Clinical studies on the therapeutic effects and the underlying mechanisms of Tyk2 inhibitors in autoimmune or chronic inflammatory diseases are currently ongoing. This review summarizes the findings of studies examining the role of Tyk2 in immune and/or inflammatory responses using Tyk2-deficient cells and mice.
Key Words: Tyrosine kinase 2; Cytokines; Signal transduction; Immune system; Inflammation
INTRODUCTION
Cytokines function as effectors and regulate the proliferation, differentiation, and functions of immune cells and consequently aid in the clearance of invading pathogens. However, cytokines are also involved in the onset and development of autoimmune diseases[1]. Cytokine-specific cell surface receptors exhibit conformational changes upon activation, which result in activation of the Janus family of protein tyrosine kinases (Jaks). Activated Jaks promote the recruitment and phosphorylation of the transcription factor signal transducer and activator of transcription (STAT).Nuclear translocation of activated STATs induces the expression of cytokineresponsive genes. Thus, the Jak-STAT pathway transduces signals from various cytokine receptor superfamily members[2-4].
The Jak family comprises Jak1, Jak2, Jak3, and tyrosine kinase 2 (Tyk2), which are activated by distinct cytokines[2-4]. Jak1 binds to interferon (IFN), interleukin (IL)-6,and IL-10 receptors that contain a common γ chain and gp130 subunit, while Jak2 binds to IL-3 and erythropoietin, growth hormone, and prolactin hormone-like receptors. Tyk2 binds to IFN, IL-12, and IL-23 receptors. Jak3, whose expression is localized to hematopoietic cells, binds exclusively to receptors that contain common γ chains along with Jak1. Moreover,Jak1deficiency in mice results in perinatal lethality and impaired lymphocyte development[5]. The embryonic lethality inJak2-deficient mice is attributed to insufficient definitive erythropoiesis[6].Jak3deficiency results in dysfunctional mature T and B lymphocytes and leads to severe combined immunodeficiency[7,8]. AlthoughTyk2-deficient mice are viable, they are susceptible to viral infections[9,10].
Previous studies using experimental models, such asTyk2-deficient mice have demonstrated that Tyk2 primarily functions in the IL-12 and IFN-α/β signaling pathways[9,10]. In humans, a mutation inTYK2, which causes an autosomal recessive form of hyper IgE syndrome (AR-HIES), affects the IL-23, IL-10, and IL-6 signal transduction pathways (Figure 1)[11]. Tyk2 is involved in both innate and acquired immunity. Here, the current knowledge on the involvement of Tyk2 in immune responses has been reviewed, and the potential clinical applications of Tyk2 inhibitors have been discussed.
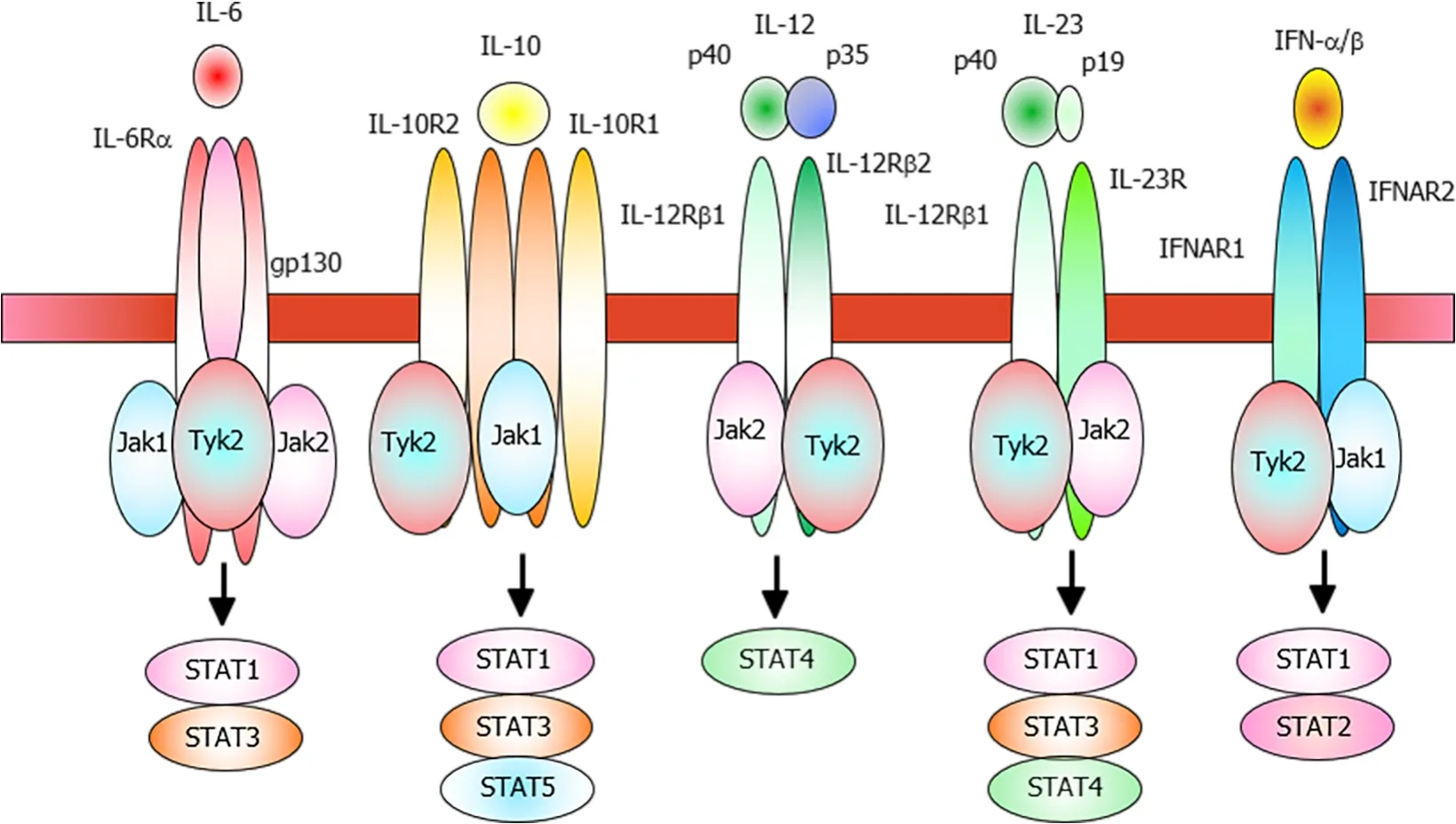
Figure 1 Schematic representation of the tyrosine kinase 2-related cytokine receptors.
ROLE OF TYK2 IN INFLAMMATORY RESPONSES
IFN system
Tyk2 was originally identified as a protein kinase that can compensate for the loss of IFN response in mutant fibroblasts[12]. IFN-α specifically activates Tyk2 and Jak1,which leads to the phosphorylation of STAT1 and STAT2 and the dimerization of activated STATs. The nuclear translocation of dimerized STATs induces the expression of target genes[3,13].
Type I IFNs are constitutively expressed in various cells, including macrophages.Although the constitutive expression of type I IFNs is low, they can regulate physiological cellular functions in an autocrine or a paracrine manner[14,15]. Tyk2 promotes the constitutive production of type I IFNs in macrophages under steadystate conditions, as well as during the innate immune responses against bacterial components. The basal and lipopolysaccharide (LPS)-induced expression levels of type I IFN are dysregulated inTyk2-deficient macrophages[16]. Moreover,Tyk2-deficient andIfnb-deficient mice are resistant to high-dose LPS-induced lethal septic shock[16,17]. Additionally, the expression of type I IFN-responsive genes, especially under steady-state conditions, was downregulated inTyk2-deficient macrophages[18].Therefore, Tyk2 is partially involved in macrophage activation by regulating autocrine and/or exogenous IFN production in the neighboring immune cells.
IL-12 and IL-23 systems
Helper T cells can be classified into the following two subsets based on their cytokine profiles: T helper 1 (Th1) and Th2 cells[19]. IL-12 and IL-4 promote the differentiation of naïve CD4+T cells into Th1 cells and Th2 cells, respectively. Heterodimeric IL-12 comprises covalently linked p35 and p40 subunits. Both IL-12 and IL-23 comprise the p40 subunit[20]. IL-23 (comprising p40 and unique p19 subunits) promotes the differentiation of Th17 cells, which secrete the effector cytokines IL-17, IL-21, and IL-22[21,22]. Th17 cells can promote enhanced inflammatory responses to eliminate microbial pathogens. However, Th17 cells are considered highly pathogenic as excessive and prolonged activation of Th17 cells can result in autoimmune and inflammatory disorders, including inflammatory bowel diseases (IBD) and rheumatoid arthritis(RA), in humans (Figure 2)[21,22].
The activation of IL-12 receptor, which is associated with Tyk2 and Jak2, activates STAT4[23,24]. Phosphorylated Stat4 along with signals from the activated T cell receptor induces the expression of T-bet, which is a master transcriptional factor for Th1 differentiation[25]. IL-23, whose receptor is associated with Tyk2, induces the proliferation, survival, and functional maturation of Th17 cells[22,26] although Th17 cell differentiation is dependent on signals from TGF-β and IL-6 (Figure 3)[22]. STAT3,a major downstream effector of the Th17-related cytokine pathway, is critical for commitment to the Th17 Lineage, whereas STAT4 and STAT6 are essential for commitment to the Th1 and Th2 Lineages, respectively[27,28]. Additionally,Tyk2-deficient macrophages do not produce nitric oxide in response to LPS stimulation[16].Tyk2-deficient dendritic cells do not produce IL-12 and IL-23 upon stimulation with CpG oligodeoxynucleotides and consequently cannot induce Th1 cell differentiation[29]. Therefore, Tyk2 is involved in the host defense response by regulating the production and function of both Th1 and Th17 cells.
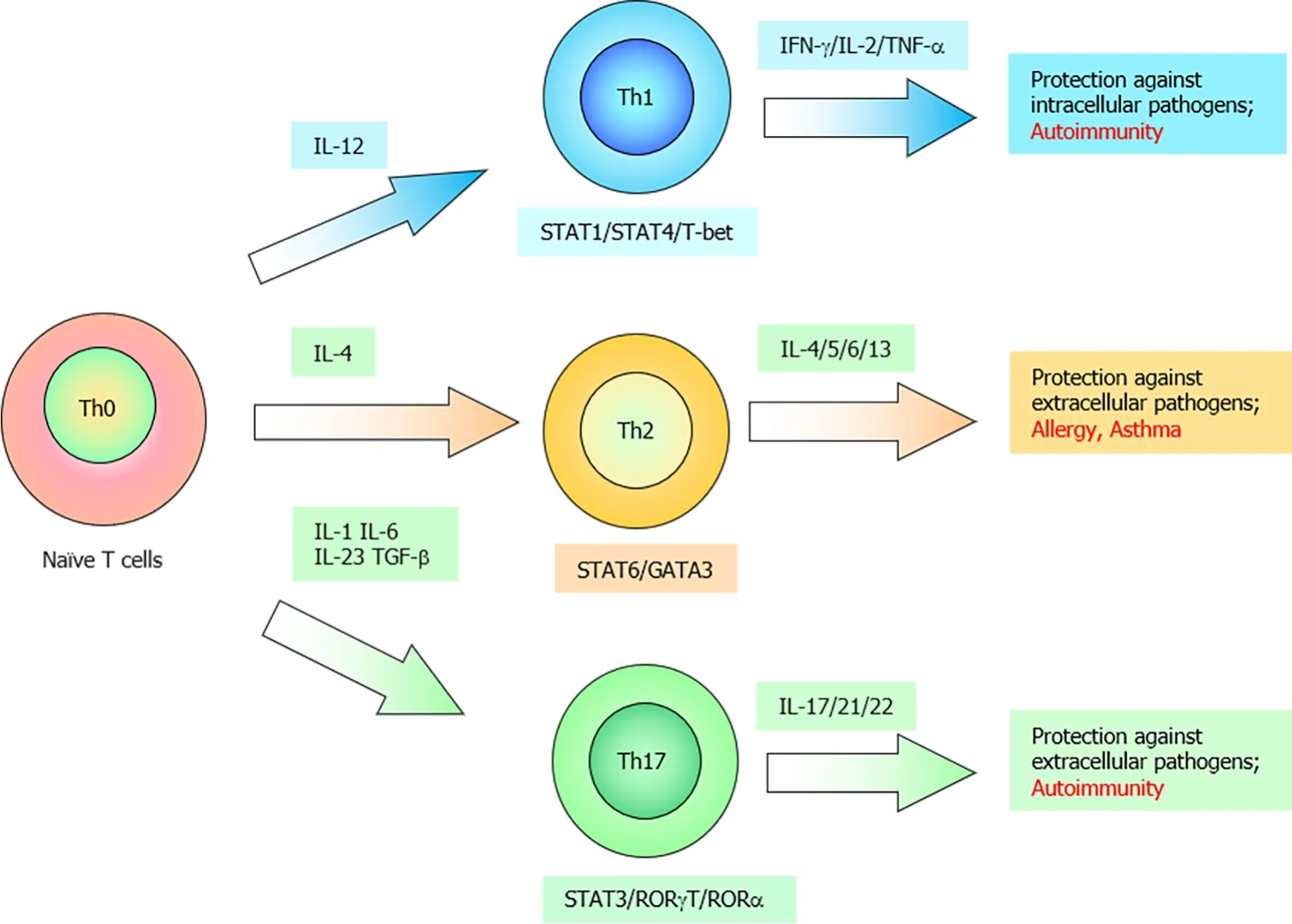
Figure 2 Schematic representation of naïve T cell differentiation into T helper 1, T helper 2, or T helper 17 cells depending on the cytokine profile.

Figure 3 Illustration of interleukin-12 and interleukin-23, as well as their receptors and downstream signaling pathways.
Inhibitory effects of type I IFNs on B lymphopoiesis are mediated through the TYK2-DAXX axis
Interactions between IFN-α and its receptor promote potent antiviral and antiproliferative activities against the target cells[3,4]. IFN-α stimulation specifically activates Tyk2 and Jak1, which leads to the phosphorylation of STAT1 and STAT2. Nuclear translocation of phosphorylated STATs (in the form of homodimers or heterodimers)promotes target gene expression[3,4].Jak1-deficient cells are not responsive to IFN-α stimulation[30], whereasTyk2-deficient cells cannot inhibit lymphocyte growth[31].Additionally,Stat1-deficient mice do not respond to IFN-α simulation[32,33], whileStat2-deficient mice are highly susceptible to viral infections[34].
Analysis of the colony forming unit (CFU) of bone marrow cells in the presence of IL-7 is a powerful tool to evaluate the growth capacity of B lymphocyte progenitors[35]. The CFU values of bone marrow cells in the presence of IL-7 were not markedly different between wild-type (WT) andTyk2-deficient mice, which indicated that Tyk2 did not affect the number of IL-7-responsive B lymphocyte progenitors under steadystate conditions[31]. IFN-α, which is a potent inhibitor of IL-7-dependent growth of B lymphocyte progenitors, effectively inhibits B lymphocyte differentiation at the pro-B cell stage[36]. The CFU values of WT bone marrow cells in the presence of IL-7 markedly decreased upon stimulation with IFN-α. In contrast, the CFU values ofTyk2-deficient bone marrow cells in the presence of IL-7 did not decrease upon stimulation with IFN-α[31]. The knockout ofTyk2completely inhibited the IFN-α -induced elevation and nuclear accumulation of death-associated protein (Daxx)[31]. Daxx was originally identified as a Fas-binding protein[37] and it plays crucial roles in the type I IFN-induced growth suppression of B lymphocyte progenitors[38]. One study used the sumoylation-defective Daxx KA mutant (Daxx K630/631A) to investigate the involvement of Daxx in decreasing the growth of Ba/F3 pro-B cells in the presence of IL-7 through IFN-α. The study demonstrated that Daxx KA is localized to the cytoplasm,whereas Daxx WT is localized to the nucleus[39]. Moreover, overexpression of Daxx KA conferred resistance to IFN-α -induced growth inhibition in a murine pro-B cell line Ba/F3. Treating Daxx KA-expressing Ba/F3 cells with leptomycin B, an exportin inhibitor, enhanced the nuclear localization of Daxx KA, and the growth of the cells was suppressed upon stimulation with IFN-α. Additionally, Daxx KA binds only weakly to promyelocytic leukemia protein (PML), which aids in the nuclear localization of Daxx. Conversely, overexpression of PML promotes the recruitment of Daxx to the PML nuclear bodies. A fusion protein comprising Daxx and a small ubiquitin-related modifier enhances the nuclear localization of Daxx and inhibits Ba/F3 cell growth. This indicates that IFN-α -induced inhibition of B lymphocyte progenitor growth requires nuclear localization of Daxx, which is dependent on sumoylation and interactions with PML. Therefore, the Tyk2-Daxx axis plays an essential role in IFN-α -induced growth inhibition of B lymphocyte progenitors.
PATHOLOGICAL SIGNIFICANCE OF TYK2 IN IMMUNE AND INFLAMMATORY DISEASES: DATA FROM MURINE EXPERIMENTAL MODELS
RA
RA is associated with joint inflammation and pain owing to a runaway immune system that elicits immune responses against the synovium of the joints of the hands,knees, or ankles. Murine experimental models for arthritis have provided useful information on various cellular and molecular mechanisms associated with RA[40].
Collagen-induced arthritis (CIA) mice are widely utilized as an experimental model for human RA[41]. Development of arthritis involves the production of autoantibodies in response to collagen and the subsequent inflammatory response against joints. Mice harboringTyk2 polymorphisms exhibit differential susceptibility to CIA[42,43].B10.Q/Ai mice are highly susceptible to CIA, whereas B10.D1 mice are resistant. This suggests thatTyk2deficiency results in the defined clinical RA.
Monitoring of the inflammatory response in the anti-type II collagen (CII) antibodyinduced arthritis (CAIA) experimental model provides useful information on the mechanisms of RA[44].Tyk2-deficient mice are highly resistant to the development of CAIA. Histological analysis has revealed thatTyk2deficiency downregulated the inflammatory cell infiltration into the synovium[45]. Additionally, the production of IFN-γ, tumor necrosis factor (TNF)-α, IL-6, and matrix metalloproteinases (MMPs) was severely impaired inTyk2-deficient mice[45]. TNF-α and IL-6, which are secreted by macrophages, function as pro-inflammatory cytokines in the CAIA model. MMPs,which are expressed in chondrocytes, synoviocytes, and macrophages, are reported to be involved in the degradation and damage of articular cartilage[46,47].Tyk2-deficient macrophages cannot produce nitric oxide in response to LPS stimulation. Meanwhile,Tyk2-deficient dendritic cells cannot produce IL-12 and IL-23 in response to CpG oligodeoxynucleotides[29]. The potential mechanisms were analyzed using the anti-CII monoclonal antibody, which induced the phosphorylation of STAT3 and STAT4 in the draining lymph node cells. Phosphorylated STAT3 and STAT4 were detected in WT but not inTyk2-deficient mice[45]. This suggests that Tyk2 promotes the production and downstream signaling of Th1/Th17-related cytokines, which are activated through STAT3 and STAT4.
Tyk2deficiency markedly decreased the susceptibility to arthritis development in both CIA and CAIA murine models, which indicated that Tyk2 plays an important role in adaptive autoimmunity and inflammatory responses. Therefore, Tyk2 regulates multiple steps involved in the onset and development of RA.
Multiple sclerosis
Multiple sclerosis (MS) is characterized by the lack of myelin, a protective sheath covering nerve fibers, which leads to disruption of the communication between the brain and other tissues[48]. Patients with MS exhibit various symptoms, such as difficulty in walking and balancing, muscle weakness and spasticity, and loss of concentration and memory. The murine experimental autoimmune encephalomyelitis(EAE) model, which is an animal model for human MS, is triggered by immunization with myelin antigens or by the adoptive transfer of myelin-specific CD4+effector cells[49].Tyk2-deficient mice exhibit decreased clinical scores and limited lymphocyte infiltration into the inflamed central nervous system[50]. The involvement of Tyk2 in EAE was confirmed using mice harboring differentTyk2polymorphisms. B10.D1 mice,which harbor theTyk2Aallele, are resistant to EAE development. The insufficient responses can be compensated by one copy of theTyk2Gallele from B10.Q/Ai mice[51].
IBD
Crohn's disease is characterized by inflammation of the digestive tract. Patients with Crohn’s disease exhibit severe diarrhea, abdominal pain, fatigue, weight loss, and malnutrition[52]. Dextran sulfate sodium (DSS)-induced colitis, a mouse model for human Crohn’s disease, is generated by supplementing mice with DSS through drinking water. The disease activity index and histological score were assessed using the combined scores of weight loss, consistency, and bleeding and acute clinical symptoms with diarrhea and/or extremely bloody stools[53]. Compared with that in WT DSS-induced colitis mice, disease development was delayed inTyk2-deficient DSSinduced colitis mice[45]. Oral supplementation of DSS activates intestinal macrophages, which leads to enhanced production of inflammatory cytokines and chemokines. Subsequently, lymphocytes are recruited to the inflammatory sites and elicit Th1 and/or Th17 responses. During this inflammatory process, Tyk2 can regulate the functions of macrophages and dendritic cells, as well as the Th1 and Th17 responses.Indeed, the mRNA levels of DSS-induced Th1 cell-related or Th17 cell-related cytokines were significantly downregulated in the colon tissues ofTyk2-deficient mice[45]. A genome-wide association study identifiedTyk2as a Crohn’s disease susceptibility locus[54].
Ulcerative colitis is characterized by inflammation and ulcers in the large intestine and rectum. Patients with ulcerative colitis exhibit diarrhea with bloody stool,abdominal pain, fever, and body weight loss[55]. To model human ulcerative colitis in mice, 2,4,6-trinitrobenzene sulfonic acid (TNBS) is used[45]. WT mice treated with TNBS die within 3 days due to the induction of massive colitis. However, approximately 50% ofTyk2-deficient mice survive after treatment with TNBS. Additionally,the bodyweight of the surviving mice returned to the physiological range after recovery from diarrhea[45].
Therefore, Tyk2 is a key molecule for the development of IBD.
Psoriasis
Psoriasis is characterized by scaly erythematous lesions in the skin, epidermal hyperplasia, parakeratosis, and accumulation of inflammatory cells[56]. The inflammatory response is mediated by several cytokines, such as TNF-α, IL-17, and IL-23.The mouse model for human psoriasis was developed by treatment with imiquimod(IMQ), a ligand for TLR7[57].Il23p19-deficient andIl17a–deficient mice exhibit decreased scores for erythema, scaling, and thickness upon treatment with IMQ, which suggests that the IL-23/Th17 axis and the Th17 cell-produced cytokines are essential for the development of skin abnormalities[57]. A genome-wide association study identifiedTyk2as a psoriasis susceptibility locus[54].Tyk2deficiency mitigates IMQinduced enhanced ear thickness, which results from epidermal hyperplasia and inflammatory cell infiltration[45].Tyk2-deficient mice exhibit markedly decreased numbers of CD4+IL-17+or CD4+IFN-γ+T cells in the draining lymph nodes and downregulated mRNA levels of Th17 cell-related cytokines upon treatment with IMQ[45].
The IL-23-induced skin inflammation mouse model is another promising model for human psoriasis[58]. In this IL-23-induced model,Tyk2-deficient mice exhibited reduced ear skin swelling, epidermal hyperplasia, Th17 and IL-22-producing Th22 cell infiltration compared with wild-type mice[45].Tyk2deficiency downregulates the production of pro-inflammatory cytokines and psoriasis-related anti-microbial peptides.
IL-23 and IL-22 coordinate to promote skin inflammation[58,59]. Tyk2-mediated signals are essential for the induction of enhanced leukocyte infiltration and inflammatory cytokine production. Enhanced keratinocyte proliferation and differentiation are highly dependent on IL-17 and IL-22. Previous studies have reported that Tyk2 directly regulates IL-22-dependent processes as evidenced by the downregulation of STAT3 phosphorylation inTyk2knockdown human keratinocyte HaCaT cells after IL-22 stimulation[45]. Therefore, Tyk2 has a critical role in the IL-22 signaling cascade that is involved in inducing epidermal hyperplasia.
IκB-ζ, an IL-17-induced protein encoded byNFKBIZ[60], is upregulated in the epidermal keratinocytes of psoriatic lesions[61].NFKBIZis located in the psoriasis susceptibility locus at 3q12.3[62]. IκB-ζ, a nuclear IκB family protein, positively or negatively modulates NF-κB-dependent and/or STAT3-dependent transcription[63-65]. Tyk2 is involved in IL-17–induced IκB-ζ expression in keratinocytes[66].Tyk2-deficient mice exhibited only slight inflammation and downregulated mRNA levels ofNfkbizupon treatment with IMQ. The catalytic activities of Tyk2 and STAT3 are required for IκB-ζ promoter activity in the HaCaT cells. The signaling pathways activated by IL-17 regulate mRNA stability[66-70]. ZC3H12A, which exhibits endoribonuclease activity, functions as a negative feedback regulator for inflammatory signaling[71-74]. The ubiquitin-proteasome pathway rapidly degrades ZC3H12A in IL-17-treated, IL-1β-treated, or IL-36–treated keratinocytes[72,74], which suggests that the stimulus-induced ZC3H12A downregulation can markedly suppress the inhibitory effects on mRNA expression.
Therefore, Tyk2 promotes the development of psoriasis by transducing IL-22 and IL-23 signals and regulating NFKBIZ along with the IL-17/ZC3H12A axis.
Sarcoidosis
Sarcoidosis is characterized by the aberrant accumulation of inflammatory cells, which typically form granulomas. Sarcoidosis usually begins in the lungs, skin, lymph nodes,eyes, heart, or other organs[75]. The murine model for human sarcoidosis is developed by intraperitoneally administering mice with heat-killedPropionibacterium acnes(P.acnes), which induces dense granulomas in the liver[76]. IL-12-IFN-γ axis is required for the induction since neitherIfngr-deficient norIl12p40-deficient mice form hepatic granulomas afterP. acnesinjection[76].Tyk2-deficient mice injected withP. acnesexhibit reduced serum IFN-γ level and decreased formation of hepatic granulomas compared with wild-type mice[45], indicating that Tyk2 has a role inP. acnes-induced granuloma formation.
Delayed-type hypersensitivity
Delayed-type hypersensitivity (DTH), which protects against various pathogens, such as mycobacteria, fungi, and parasites, contributes to transplant rejection and tumor immunity[77]. DTH is mainly dependent on T cells and develops 24–72 h after exposure to a foreign antigen. The DTH response analysis is based on a Th1/Th17 type model as the hypersensitivity response is defective inIl12p40-deficient andIl23p19-deficient mice[78]. The sensitization phase is triggered by immunizing mice with a specific protein antigen (methylated BSA). The elicitation phase, which is initiated by the second injection of methylated BSA into the rear footpad of the pre-immunized mice, results in footpad swelling. Footpad swelling was significantly alleviated inTyk2-deficient mice, which indicated the role of Tyk2 in DTH responses[45].
HIES
Tyk2 AR-HIES is a hereditary (autosomal recessive) disease involving aTyk2mutation[79]. Patients with Tyk2 AR-HIES are characterized by repeated viral and mycobacterial infections, atopic dermatitis, and enhanced levels of IgE[11]. Therefore, Tyk2 may have a broader and more important role in immunological responses than expected from studies conducted usingTyk2-deficient mice.
POTENTIAL CLINICAL APPLICATIONS OF TYK2 INHIBITORS
The firstin vivoevidence for the roles of Jaks in cytokine signaling originated from a human case study of severe combined immunodeficiency. Mutations in Jak3 or its receptor (a common g cytokine receptor chain) were detected in this case[80,81].Another example is a somatic Jak2 valine-to-phenylalanine mutation (V617F), which is detected in more than 90% of the patients with polycythemia and some patients with essential thrombocythemia and primary myelofibrosis[82]. Activating point mutations inJak1are detected in DNA samples from patients with acute lymphoblastic leukemia and are rarely observed in patients with acute myeloid leukemia[83]. Thus, dysregulation of the Jak-mediated signaling pathway is associated with the pathogenesis of different diseases, including hematological malignancies, autoimmune diseases, and immune-disrupted conditions. Studies onTyk2-deficient mice or human patients with mutatedTyk2alleles have revealed that Tyk2 is a key player in the pathogenesis of autoimmune and/or inflammatory diseases.
Imatinib, a Bcr-Abl kinase inhibitor, exerts potent therapeutic effects in patients with chronic myelocytic leukemia[84]. Hence, various kinase inhibitors with strict selectivity and potency have been developed[85]. Jak inhibitors exert potent therapeutic effects by mitigating high levels of circulating immune/inflammatory cytokines. These results strongly suggest that Tyk2 is a potential therapeutic target for patients with immune and/or inflammatory diseases.
First-generation Jak inhibitors typically target two or three Jak types. Therefore,first-generation Jak inhibitors are associated with broader effects and more adverse events than the new-generation drugs, which specifically target one Jak type.Currently, several Jak inhibitors are used to treat various human diseases[86]. For example, ruxolitinib, an inhibitor of Jak1 and Jak2, has been approved to treat patients with myelofibrosis and polycythemia vera[87]. Tofacitinib, an inhibitor of Jak1, Jak2,and Jak3, has been approved to treat patients with RA, psoriatic arthritis, and ulcerative colitis[88]. Baricitinib, an inhibitor of Jak1 and Jak2, is used to treat patients with RA[89]. In methotrexate-inadequate responders, both tofacitinib and baricitinib provided enhanced therapeutic responses in patients with RA when compared with placebo[90]. In a phase 2 trial involving patients with psoriasis, the response rate to deucravacitinib (BMS-986165), a Tyk2 selective inhibitor[91], was significantly higher than that to placebo after 12 wk of administration. Treatment with BMS-986165 did not affect the blood cell counts or the serum levels of liver enzymes, lipids, and creatinine.However, BMS-986165 was associated with some severe adverse effects, such as malignant melanoma. Theoretically, therapeutic strategies targeting the immune system may increase the risk of infections from various pathogens, such as herpes zoster virus, cytomegalovirus, and Epstein–Barr virus. Therefore, further studies are needed to determine the long-term efficacy and safety of Tyk2 inhibitors. Additionally,clinical trials on Tyk2 inhibitors will aid in devising better therapeutic strategies for immune/inflammatory diseases than the currently marketed therapeutics.
CONCLUSION
This review summarized the involvement of Tyk2 in the immune system and its possible potential roles in the onset and development of immune and inflammatory diseases (Figure 4).

Figure 4 Schematic representation of the involvement of tyrosine kinase 2 in immune and inflammatory responses and its pathological significance.
Studies on theTyk2-deficient cells have revealed the involvement of the IFN system and IL-12/IL-23 axis. Constitutive production of a small amount of type I IFNs elicits a pro-inflammatory response against the invading pathogens and mitigates aberrant inflammation by promoting the expression of IL-10, a potent anti-inflammatory cytokine. Tyk2 is critical for maintaining the basal levels of IFNs. The IFN-α-induced decreased CFUs of bone marrow cells in the presence of IL-7 were also dependent on Tyk2. Tyk2 contributes to IFN-α signaling by promoting the nuclear translocation of Daxx and the formation of the Daxx/PML complex, which leads to growth inhibition.Additionally, Tyk2 interacts with the receptors for type I IFN, IL-6, IL-10, IL-12, and IL-23 (Figure 1). Moreover, Tyk2 is essential for IL-12-induced differentiation into Th1 cells, as well as IL-23-induced proliferation, survival, and functional maturation of Th17 cells. Additionally, Tyk2 mediates the production of nitric oxide in macrophages and IL-12 and IL-23 in dendritic cells after the invasion of pathogens.
Studies usingTyk2-deficient mice have revealed the potential involvement of Tyk2 in the onset and development of various immune and/or inflammatory disorders,such as RA, MS, IBD, psoriasis, sarcoidosis, and DTH. The development of most phenotypes in these models was mediated by Th1 and Th17 cells, whose differentiation and functions are highly dependent on Tyk2. Additionally, Tyk2 contributes to IL-17-induced IκB-ζ expression in IMQ-induced skin inflammation.
The experimental data summarized in this review along with the known clinical success of the novel Jak inhibitors indicate the therapeutic potential of Tyk2 inhibitors in the clinical setting. Further clinical trials are needed to examine the safety and efficacy profiles of Tyk2 inhibitors for treating psoriasis. Additionally, Tyk2 inhibitors are likely to be widely approved for various Th1/Th17-related immune/inflammatory diseases.
