超声波提取-固相萃取-液相色谱-串联质谱法同时测定沉积物中24种皮质类固醇激素
2022-02-14周永顺杨可欣林粲源吴翠琴张舒寒
周永顺, 龚 剑, 杨可欣, 林粲源, 吴翠琴, 张舒寒
(广州大学环境科学与工程学院, 珠江三角洲水质安全与保护教育部重点实验室, 广东省放射性核素污染控制与资源化重点实验室, 广东 广州 510006)
皮质类固醇激素(corticosteroids, CSs)是一类由肾上腺皮质所合成的重要激素,几乎参与了人类和动物的所有生命活动[1,2]。人工合成的CSs类药物被广泛应用于炎症、免疫类疾病治疗[3,4],但长期使用可能导致人体免疫力下降、肥胖等疾病[5]。CSs可通过人体和动物的排泄、污水处理厂的不完全处理排放,并最终进入天然水环境中。相关研究表明多种CSs普遍存在于环境水体中,可能已对水生系统造成不利影响[6,7]。与其他有机污染物一样,CSs可通过吸附作用附着于悬浮颗粒上并随之迁移,最终沉积到水底,富集于沉积物中;也可在水动力或其他理化条件变化时重新释放到水体,并通过生物富集和食物链的传递作用进而危害生态系统和人类健康[8]。迄今为止,关于环境中CSs污染现状的研究主要集中在地表水和污水处理厂上[9,10],而对于其在沉积物等天然固相介质中的赋存状况则鲜有报道[11]。
环境中CSs的残留水平不高(μg/L至ng/L级),沉积物样品基质组成复杂,前处理难度较大,因此,目前关于沉积物中CSs的残留分析方法仍十分有限。有关CSs在天然环境中的含量水平、污染特征等研究刚起步[9-11],对该类污染物环境地球化学行为的认识亦非常有限。这主要受制于缺少相关的分析方法,特别是缺少针对基质复杂的环境介质中CSs多残留的痕量分析方法。Zhang等[12]测定了基质较为复杂的市政污水中3种糖皮质激素,然而该方法回收率较低,检出限和定量限较高,且实际样品中未能检测出目标物;Liu等[13]和Chen等[14]分析了水体颗粒物和沉积物中4~5种糖皮质激素,仅有皮质醇被检出。虽然之前Liu等[15]、杨雷等[16]、Fan等[17]、Weizel等[18]开展了沉积物和污泥中数十种类固醇激素的监测,但就CSs而言该类分析方法缺乏针对性和系统性。而且其目标物仅包含5~12种糖皮质激素,不仅未涉及盐皮质激素,也未能覆盖大多常见常用的CSs。水环境中沉积物既是污染物的“汇”也是“源”,因此,有必要针对性地发展一套简便、灵敏的沉积物中CSs多残留的痕量分析方法,并将其应用到实际的环境监测中,为更系统地研究CSs在环境多介质中的赋存状况和环境行为提供技术保障。
针对基质更为复杂的环境沉积物,本研究在水样分析方法[19]的基础上,进一步采用超声波辅助提取联合固相萃取(SPE)技术对样品进行前处理,并对提取液开展二次深度净化降低基质干扰;重新对质谱条件进行优化,兼顾信号响应和仪器耐受性。最终建立了一套利用超高效液相色谱-串联质谱(UPLC-MS/MS)同时检测沉积物中24种CSs的定量分析方法,并应用于珠江沉积物的CSs测定。
1 实验部分
1.1 仪器、试剂与材料
1260 Infinity-6460 QQQ超高效液相色谱-三重四极杆质谱联用系统(UPLC-MS/MS, Agilent公司)及Agilent MassHunter定量分析软件;Autotrace280全自动固相萃取仪(美国Thermo公司); Rotavapor R-120旋转蒸发仪(瑞士BUCHI公司); EFAA-DC12氮吹仪(上海安谱公司); Oasis HLB SPE柱(500 mg, 6 mL, Waters公司); Silica SPE柱(500 mg, 6 mL, Waters公司); LC-NH2SPE柱(500 mg, 3 mL, Supelco公司)。
24种CSs标准样品(见表1)均购自美国Sigma-Aldrich公司,纯度均大于98%; 5种同位素替代物标准品:氢化可的松-d4、地塞米松-d4、曲安奈德-13C3、布地奈德-d8、丙酸氟替卡松-d5均购自加拿大Toronto Research Chemicals公司,纯度均大于95%;甲醇、乙酸乙酯、乙腈、丙酮(HPLC级,德国CNW公司);实验用水均为超纯水(电阻率18.25 MΩ·cm)。
实际样品为珠江三角洲河流表层沉积物。
1.2 标准溶液的配制
首先称取适量标准品,用甲醇配制成质量浓度为100 mg/L的单标储备液。取适量储备液用甲醇稀释配制成10 mg/L的24种目标物的混合标准溶液和10 mg/L的5种同位素替代物混合标准溶液。然后用甲醇逐级稀释至质量浓度为1.0~100 μg/L的系列混合标准工作溶液;另取适量的5种同位素替代物混合标准溶液稀释成1 mg/L的内标工作液备用。所有标准溶液置于冰箱-20 ℃避光储存。
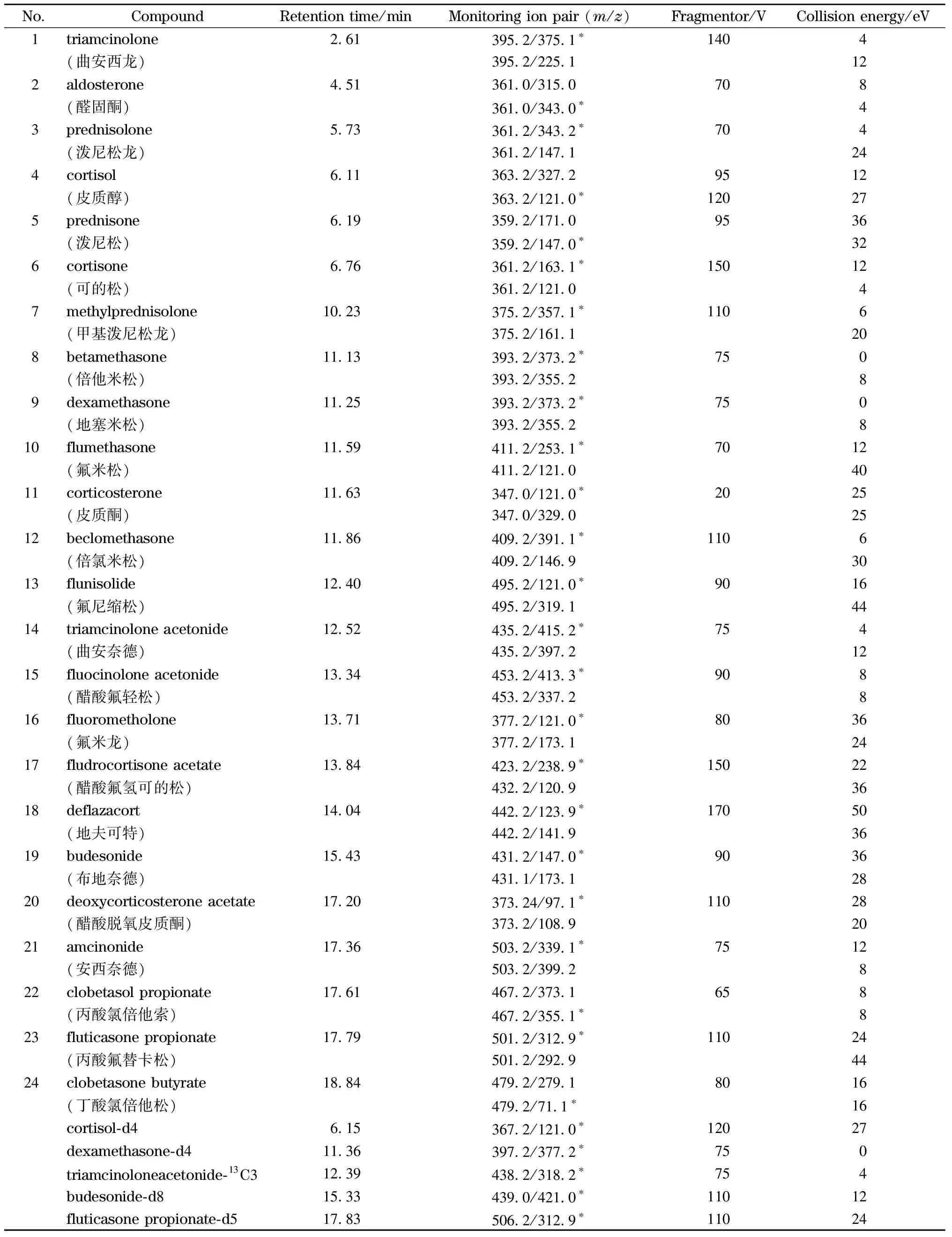
表 1 24种目标化合物的质谱参数Table 1 Mass spectrometric parameters for the 24 target compounds
1.3 样品的采集与处理
用不锈钢抓斗式采样器从珠江三角洲河流采集表层(0~20 cm)沉积物样品,转移至实验室并置于-20 ℃冷冻保存至分析。
称取2.0 g经冷冻干燥、研磨过筛的沉积物样品置于50 mL具塞聚丙烯离心管中,加入0.5 g铜片、10 mL甲醇-丙酮(1∶1, v/v)、20 μL的内标工作液,混匀后超声提取10 min,再以6 000 r/min速度离心10 min并收集上层清液。上述步骤重复3次并合并提取液。利用旋转蒸发仪将提取液浓缩至1 mL后,溶于200 mL超纯水中,溶液用于固相萃取。Oasis HLB小柱依次用6 mL乙酸乙酯、6 mL乙腈、12 mL超纯水活化后,使用全自动固相萃取仪以10 mL/min的流速将试样溶液过柱。上样完毕后,先用10 mL 10%(v/v)乙腈水溶液淋洗,接着氮吹干燥柱床,然后依次用6 mL乙酸乙酯-乙腈(1∶1, v/v)、6 mL乙酸乙酯洗脱。收集的洗脱液经旋转蒸发、氮气吹干后复溶于1 mL甲醇。用5 mL乙酸乙酯、5 mL甲醇活化LC-NH2小柱,再将甲醇浓缩液过柱净化,用5 mL甲醇洗脱。用氮气将洗脱液吹至近干并以甲醇定容至0.5 mL,待UPLC-MS/MS测定。

图 1 提取试剂对24种皮质类固醇激素回收率的影响(n=5)Fig. 1 Effect of extracting solvents on recoveries of the 24 CSs (n=5)
1.4 色谱条件
Agilent ZORBAX Eclipse Plus C8色谱柱(100 mm×2.1 mm, 1.8 μm);流动相A: 0.1%(v/v)乙酸水溶液,流动相B:乙腈;柱温30 ℃;流速0.3 mL/min;进样量5 μL;梯度洗脱程序[19]: 0~8.0 min, 28%B; 8.0~8.1 min, 28%B ~ 40%B; 8.1~12.0 min, 40%B; 12.0~12.1 min, 40%B~60%B; 12.1~16.0 min, 60%B~70%B; 16.0~16.5 min, 70%B~100%B; 16.5~20.5 min, 100%B。
1.5 质谱条件
离子源:电喷雾电离源,正离子扫描模式(ESI+);雾化器压力:276 kPa;脱溶剂气流速:11 L/min;离子源温度:300 ℃;毛细管电压4 000 V;采集时间窗口(delta retention time)为1.2 min,采用动态多反应选择性监测扫描模式(DMRM)。优化后的24种目标化合物的质谱参数见表1。
定量分析:为了有效监控实验过程的系统误差,消减基质效应的影响,提高定量结果的准确性,本研究采用内标法定量。使用5种同位素替代物作为内标对24种目标物进行定量分析:皮质醇-d4(No.1~6)、地塞米松-d4(No.7~12)、曲安奈德-13C3(No.13~18)、布地奈德-d8(No.19~20)、丙酸氟替卡松-d5(No.21~24)(括号中为其对应的表1中目标物的编号)。
2 结果与讨论
2.1 提取溶剂的选择
选择合适的萃取溶剂能够提高目标物的提取效率同时减少杂质干扰。考虑到目标化合物种类较多、极性范围较大且沉积物样品基质复杂等特点,本研究选取弱、中、强极性的3种常用溶剂乙酸乙酯、乙酸乙酯-乙腈(1∶1, v/v)以及甲醇-丙酮(1∶1, v/v),用于样品超声萃取并比较其提取效率。
3种溶剂萃取加标样品的回收率结果见图1。可以看出,样品经乙酸乙酯溶剂提取后,所有目标物均被检出,回收率为8.58%~154.4%,相对标准偏差(RSD)为1.28%~40.8%;乙酸乙酯-乙腈(1∶1, v/v)的提取液中未能检测出曲安西龙,其余目标物回收率为6.19%~172.2%,RSD为0.64%~21.3%;样品经甲醇-丙酮(1∶1, v/v)提取后所有目标物均被检出,回收率为71.7%~130.2%,RSD为1.1%~9.7%。与前两者相比,甲醇-丙酮(1∶1, v/v)作为提取溶剂具有较高的回收率和重复性。因此本研究选择甲醇-丙酮(1∶1, v/v)作为超声萃取溶剂。
2.2 SPE净化条件的选择及优化
沉积物样品基质复杂,其提取液中难免含有大量的干扰物,不但影响仪器分析的准确性,还会缩短分离柱的使用寿命[20]。因此为尽量降低基质效应的影响,本研究考察了Silica柱和LC-NH2柱对SPE浓缩液作进一步净化的效果。其中Silica柱是以硅胶为基质的极性吸附剂,主要用于分离非水溶液中的低极性组分[21],通过增加极性高的洗脱溶剂将结构相似的化合物分开;LC-NH2柱是以氨丙基为基质的中等极性吸附剂,能够从极性溶液中萃取非极性化合物,与Silica柱一样适用于分离异构体[22]。

图 2 SPE净化条件对24种皮质类固醇激素回收率的影响(n=5)Fig. 2 Effect of SPE purification conditions on recoveries of the 24 CSs (n=5)
基于先前的研究[9],采用Oasis HLB SPE柱对样品提取液进行富集并用10 mL的10%乙腈水溶液洗柱,作为第一次净化。接着再分别用上述两种净化柱对HLB柱洗脱液进行第二次净化:(1)洗脱液浓缩后溶于1 mL乙酸乙酯-正己烷(1∶9, v/v),并转移至活化的Silica柱,并用3 mL乙酸乙酯-正己烷(1∶9, v/v)淋洗,最后用4 mL乙酸乙酯-甲醇(95∶5, v/v)洗脱,洗脱液经旋转蒸发、温和氮气吹干后用甲醇定容,待测;(2)洗脱液浓缩后溶于1 mL甲醇,并转移至活化的LC-NH2柱,用5 mL甲醇洗脱LC-NH2柱,洗脱液经旋转蒸发、温和氮气吹干,甲醇定容后待测。通过比较只经过HLB柱一次净化以及分别再经Silica柱、LC-NH2柱二次净化的3种情况,对净化效果进行综合评估。
3种净化条件下目标物回收率结果如图2所示。样品仅经过HLB柱一次净化时,目标化合物的回收率普遍偏低(25.5%~95.9%)且RSD范围较大(11.1%~42.0%),重现性较差。浓缩液经Silica柱二次净化后,目标物之一曲安西龙并未检出,可能与其在硅胶柱中保留较强,不易被洗脱下来有关。其余目标化合物的回收率为52.7%~94.7%, RSD为0.9%~8.6%。浓缩液经LC-NH2柱二次净化后,所有目标化合物均被检出,回收率为61.4%~118.4%, RSD为0.8%~6.5%。可见其净化效果优于前两种情况且回收率高、重现性较好。对比浓缩液经过LC-NH2柱净化前后代表性化合物的信号响应(见图3)发现,净化后的样品中基质干扰明显降低,目标物信号增强。因此本研究选用LC-NH2柱对样品进行二次净化。
2.3 质谱条件的优化
CSs是典型的甾体类化合物,其基本结构如图4所示。化合物在C-17、C-20和C-21处存在酮基或羟基,能够在ESI-模式下离子化,产生的母离子为[M+HCOO]-;在C-3处存在共轭羰基等多电子基团,所以在ESI+模式下能形成[M+H]+母离子。因此CSs可以在正、负两种电离模式下检测到。通过单标溶液进样,分别对24种目标物的质谱参数进行优化。首先,通过母离子的全扫描(full scan)确定化合物的准分子离子,然后在选择离子监测(SIM)模式下优化碎裂电压和毛细管电压,使[M+H]+或[M+HCOO]-响应达到最大;然后对母离子作子离子全扫描(product ion scan),选择2个具有更高丰度的特征碎片离子,优化碰撞能量以使响应最大化,最后获得响应最佳的两对MRM离子对和相应的质谱参数。

图 3 氟尼缩松、醋酸氟氢可的松、皮质酮、丙酸氯倍他索和丁酸倍他松经LC-NH2 SPE柱净化前、后的MRM色谱图Fig. 3 MRM chromatograms of flunisolide, fludrocortisone acetate, corticosterone, clobetasol propionate and clobetasone butyrate before and after purified by LC-NH2 cartridges

图 4 皮质类固醇激素的结构骨架Fig. 4 Structural features of corticosteroids

图 5 24种目标物混合标准溶液(20 μg/L)在(a)正离子模式、(b)负离子模式、和(c)正负离子模式下的总离子色谱图Fig. 5 Total ion chromatograms (TIC) of the 24 target compounds at mass concentration of 20 μg/L in (a) positive ion mode, (b) negative ion mode, and (c) positive and negative ion mode For peak Nos., see Table 1.
分别在ESI-、ESI+和ESI±模式下,对24种目标物的特征离子进行扫描。结果发现,在ESI-模式下,皮质醇、泼尼松、皮质酮等十几种目标物的信号强度过低[23];在ESI+模式下的大部分化合物基峰的相对丰度比ESI±模式高出2~6倍(见图5)。另外,考虑到ESI±模式下须使用导电毛细管,与ESI+模式下使用的石英毛细管相比,其耐脏程度低、使用寿命也较短。因此,本研究选择石英毛细管并在ESI+下对目标化合物进行分析。
2.4 基质效应评价
沉积物样品基质复杂,进行质谱分析时易受到基质干扰,影响方法的灵敏度和重现性。本研究采用向沉积物提取液和纯溶剂中加标的方式评价基质效应(ME)的影响,计算公式如下[24]:ME=(1-扣除本底后基质溶液中目标物的响应值/纯溶剂中相应目标物的响应值)×100%。
结果(见表2)发现,沉积物样品在未经LC-NH2柱净化的条件下,目标物受基质干扰的影响大,ME值范围在-7.7%~74.5%之间,大部分化合物表现为较强的基质抑制作用;利用LC-NH2柱对沉积物样品进行净化后,ME值范围为-7.9%~27.9%,大多数目标物基质效应在±20%以内,表明该净化步骤可有效降低基质干扰。为进一步消减基质效应的影响,本研究采用同位素内标法定量。这不但能有效监控分析过程,消除系统误差,也保证了定量结果的准确度。
2.5 标准曲线的线性方程、检出限和定量限
配制1.0~100 μg/L的系列混合标准溶液,同时加入5种同位素内标,质量浓度均为10 μg/L。按照浓度由低到高依次进样,以待测物与内标物定量离子的峰面积比值为纵坐标,待测物的质量浓度(μg/L)为横坐标,分别绘制标准曲线。24种CSs具有良好的线性关系,相关系数(r2)均大于0.995。按信噪比(S/N)=3和S/N=10计算方法的检出限(LOD)和定量限(LOQ),所有化合物的LOD和LOQ分别在0.14~1.25 μg/kg和0.26~2.26 μg/kg范围内(见表2)。
2.6 加标回收率和精密度
依据目前有限的报道[11],沉积物中CSs的含量约为类固醇雌激素的几倍至数十倍,而珠江流域沉积物中的类固醇雌激素残留普遍在几个μg/kg水平[25]。因此,本文分别设定了5、20、50 μg/kg 3个加标水平,取2 g沉积物并添加不同量的混合标准溶液,每个加标水平重复测定5次。结果如表3所示,样品平均加标回收率(n=5)为64.9%~125.1%, RSD(n=5)为0.4%~12.6%,均满足环境样品有机物痕量检测要求。

表 2 24种目标化合物的线性方程、相关系数、检出限、定量限和基质效应Table 2 Linear equations, correlation coefficients (R2), limits of detection (LODs), limits of quantification (LOQs) and matrix effects (MEs) for the 24 target compounds
2.7 实际样品分析
应用所建立的方法分析了3份取自珠江三角洲河流的沉积物样品,24种CSs测定结果见表4。样品中共有11种目标物(曲安西龙、倍他米松、地塞米松、曲安奈德、醋酸氟轻松、氟米龙、氟尼缩松、醋酸脱氧皮质酮、丙酸氯倍他索、丙酸氟替卡松和丁酸氯倍他松)被检出,含量范围为1.25~29.38 μg/kg。

表 4 珠江三角洲河流沉积物样品中的皮质类固醇激素含量Table 4 Contents of corticosteroids in the sediment sample from the rivers of Pearl River Delta μg/kg
3 结论
本研究建立了一种基于超声波提取-固相萃取技术对沉积物样进行前处理,应用UPLC-MS/MS同时检测沉积物中24种CSs的分析方法。该方法具有灵敏、准确和重现性好等特点,满足对沉积物中多种天然和合成CSs的痕量监测要求,可广泛应用到该类污染物环境行为和生态风险的研究中。
