微波法制备猕猴桃生物质碳点及应用于金银花中Fe3+的检测
2022-02-11赵鸿宾冷天翠刘晓梦姜艳萍李春荣孟铁宏胡先运
赵鸿宾,冷天翠,刘晓梦,姜艳萍,李春荣,孟铁宏, ,胡先运,3,
(1.黔南民族医学高等专科学校药学系,贵州都匀 558000;2.贵州医科大学第三附属医院药房,贵州都匀 558000;3.贵州省中国科学院天然产物化学重点实验室,贵州贵阳 550002)
碳点(Carbon Dots,CDs)是以碳为骨架结构的新型纳米材料,是一种分散的粒径小于10 nm的类球形纳米颗粒[1]。CDs既具有优良的荧光特性和水溶性,又具有生物相容性好[2]、化学稳定性高[3]、耐光漂白性强[4]等优点,被广泛应用于光电[5]、安全与识别[6]、离子及药物分子检测[7-11]、荧光传感[12]、细胞成像[13-14]等领域。
CDs的合成方法主要归纳为“自上而下(Topdown)”和“自下而上(Bottom-up)”两种方法[15]。自上而下法是通过氧化物切割碳而获得如碳棒、碳纤维、碳纳米管等材料。自下而上合成法则是具有-OH、-COOH、-C=O和-NH2等基团的小分子、聚合物在高温下脱水并进一步碳化制备荧光碳点。自上而下的合成方法通常有电弧放电法[16]、激光烧蚀法[17]、电化学法[18]。自下而上的合成方法通常有:溶剂热合成法[19]、燃烧法[20]、热解法[21-22]和微波合成法[23]等。其中,微波法制备CDs的操作简单,周期短,产率高,能耗低,是快速制备CDs的首选方法[24]。生物质资源因具有来源丰富、分布广泛、取材简单、成本低廉、毒性小[25]等优点,是制备CDs的理想碳源。利用生物质制备的CDs荧光性能强,安全性高,易于功能化,因此生物质CDs具有更广阔的应用空间[26-27]。目前,以果蔬为碳源合成CDs的研究已有诸多报道,杨克琴[28]以胡萝卜为生物质碳源,采用水热法合成荧光生物质碳点,并成功应用于人膀胱癌活细胞荧光成像。穆海峰等[29]以紫甘蓝为原料,硼氢化钠为还原剂,采用水热合成法制备了碳量子点。猕猴桃为常见水果,原料易得,果肉多汁,富含糖类、维生素、氨基酸等多种有机物,是合成CDs的优质碳源。
本文拟以猕猴桃为生物质碳源,乙二胺为表面修饰剂,通过正交试验优化微波法制备氮掺杂CDs的参数。初步探讨Fe3+对猕猴桃生物质CDs荧光猝灭作用,尝试建立一种测定金银花等中药材中铁含量的方法。
1 材料与方法
1.1 材料与仪器
猕猴桃 购于都匀市水果超市,经王传明副教授鉴定为美味猕猴桃品种;金银花 购于都匀市湘君大药房;乙二胺、硫酸奎宁、醋酸、醋酸钠及其它无机化学试剂 均为分析纯;实验用水 为双蒸水。
JEM-2100透射电子显微镜 日本电子株式会社;icAN9型傅立叶红外光谱仪 天津市能谱科技有限公司;F-7000荧光光谱仪 日本日立公司;Cary 100型双光束紫外可见光谱仪 北京安捷伦公司;S-2F型pH计 上海雷磁仪器公司;XT-9912型微波消解仪 上海新拓分析仪器科技有限公司;P70F23PG5(S0)型微波炉 广东格兰仕微波炉电器公司。
1.2 实验方法
1.2.1 CDs制备单因素实验
1.2.1.1 乙二胺用量 将新鲜猕猴桃去皮榨汁,经减压过滤得猕猴桃汁约200 mL,置于2~6 ℃冰箱储存备用。取一系列100 mL锥形瓶,依次加入1.0 mL猕猴桃汁(含干物质约0.13 g)和7.0 mL蒸馏水,再分别加入1、2、3、4、5、6 mL乙二胺,涡旋混匀后置于微波炉转盘中,调节功率至560 W(中高火),加热15 min。反应后取出放冷,加入适量蒸馏水溶解,转移至一系列100 mL容量瓶中加水稀释并定容。取适量溶液离心15 min(4000 r/min),取上清液经0.22 μm滤膜过滤。精密量取滤液1.0 mL稀释定容至100 mL,静置15 min,在365 nm激发光下检测其荧光强度。
1.2.1.2 加热时间 取一系列100 mL锥形瓶,依次加入1.0 mL猕猴桃汁、1.0 mL乙二胺和7.0 mL蒸馏水,涡旋混匀后置于微波炉转盘中,调节功率至560 W,逐一加热,时间分别为5、10、15、20、25、30 min。反应后取出放冷,并按1.2.1.1项下方法进行稀释、离心、过滤等处理,检测其荧光强度。
1.2.1.3 加热功率 取一系列100 mL锥形瓶,依次加入1.0 mL猕猴桃汁、1.0 mL乙二胺和7.0 mL蒸馏水,涡旋混匀后逐一放入微波炉转盘中,分别调节功率为125 W(低火)、260 W(解冻)、400 W(中火)、560 W(中高火)、700 W(高火),加热15 min。反应后取出放冷,并按1.2.1.1项下方法进行稀释、离心、过滤等处理,检测其荧光强度。
1.2.1.4 加水量 取5个100 mL锥形瓶,依次加入1.0 mL猕猴桃汁和1.0 mL乙二胺,再分别加入3、5、7、10、15 mL蒸馏水,按照1.2.1.1项下方法加热反应,并进行后续处理,检测其荧光强度。
1.2.2 正交试验设计 通过对单因素实验结果的直观分析,分别确定乙二胺用量、微波加热时间、加热功率、加水量等4个因素及其3个水平(如表1所示)。按照L9(34)正交试验设计完成CDs的制备,测定其荧光强度,并利用分析软件进行数据处理。根据正交试验优化的最佳制备条件制备猕猴桃碳点,加入适量蒸馏水溶解,稀释成100 mL CDs储备液。取20 mL CDs储备液,离心15 min(4000 r/min),取上清液经0.22 μm滤膜过滤,滤液透析12 h,减压浓缩,干燥,得53.9 mg猕猴桃碳点。

表1 正交试验因素水平表Table 1 Factors and levels table of orthogonal experiment
1.2.3 CDs结构表征 将制备的CDs按要求进行减压干燥处理,采用JEM- 2100 透射电子显微镜观察碳量子点形貌粒径;采用icAN9傅里叶变换红外光谱仪测定样品的红外光谱图,分辨率为4 cm-1,波数400~4000 cm-1;采用F-7000荧光光谱仪测定荧光发射光谱;使用Cary100型双光束紫外可见分光光度计测定样品的紫外-可见吸收光谱。
1.2.4 荧光量子产率测定 将硫酸奎宁溶于浓度为0.1 mol/L的硫酸中,制得硫酸奎宁溶液(Φs=54.0%,365 nm),作为标准参照物。在365 nm波长下,分别测定一定浓度的猕猴桃碳点溶液和硫酸奎宁溶液的紫外吸光度及荧光发射峰积分面积,并在室温条件下测定其的折光率。根据公式(1)计算荧光量子产率[30]:
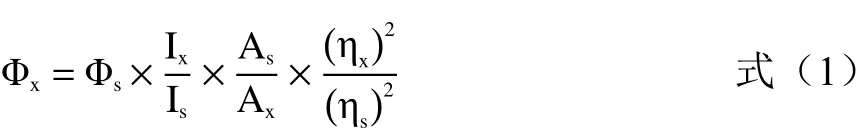
式中,Φ:荧光量子产率(%);I:荧光发射峰积分面积(a.u.);A:激发波长处溶液的吸光度;η:溶剂的折射率(%);s:硫酸奎宁标准物;x:猕猴桃生物质碳点溶液。
1.2.5 Fe3+检测条件的优化
1.2.5.1 放置时间 精密称取1.2.2项下CDs适量,加蒸馏水配制成2.7 μg/mL的CDs溶液。取CDs溶液1.0 mL置于10 mL容量瓶中,加入0.01 mol/L的FeCl3溶液1.0 mL,分别于5、10、15、20、30、45 min测其荧光强度,考察CDs与Fe3+放置时间。
1.2.5.2 缓冲溶液pH 取一系列10 mL容量瓶,依次加入1.2.4.1项下CDs溶液1.0 mL、0.01 mol/L的FeCl3溶液1.0 mL,再分别加入pH4.2、4.6、5.0、6.0的HAc-NaAc缓冲溶液1.0 mL,加去离子水稀释至刻度,充分混合后静置30 min,检测荧光强度,比较在不同pH的HAc-NaAc缓冲溶液中,Fe3+对CDs溶液荧光猝灭程度。
1.2.6 CDs应用于Fe3+的检测 依据1.2.4项下优化条件,在一系列10 mL容量瓶中,加入1.2.4.1项下浓度为2.7 μg/mL的CDs溶液1.0 mL、HAc-NaAc缓冲溶液(pH4.6)1.0 mL和不同体积的0.01 mol/L FeCl3溶液,加水稀释至刻度,摇匀,静置30 min,测定荧光强度,考察Fe3+检测的线性范围。
另取一系列10 mL容量瓶,分别加入CDs溶液1.0 mL、HAc-NaAc缓冲溶液(pH4.6)1.0 mL和0.01 mol/L不同离子的盐溶液1.0 mL,加水稀释至刻度,摇匀,静置30 min,空白试验平行操作,检测其荧光强度,考察该检测方法的选择性。
1.2.7 金银花样品的处理及其Fe3+的检测
1.2.7.1 金银花样品前处理 精密称取金银花粉末0.1 g,置于聚四氟乙烯消解罐中,先加入4 mL浓硝酸,放置30 min,再加入2 mL H2O2,加盖放入微波消解仪中按表2程序消解。待消解完成将消解罐置于90 ℃电热板上加热,将酸赶尽,冷却后收集于100 mL容量瓶中,并用超纯水定容至刻度,即得金银花供试品溶液,置于2~5 ℃冰箱中保存备用。

表2 微波消解程序Table 2 Program of microwave digestion
1.2.7.2 金银花样品中Fe3+的检测 在10 mL容量瓶中,加入1.2.4.1项下同浓度的CDs溶液1.0 mL、HAc-NaAc缓冲溶液(pH4.6) 1.0 mL和1.0 mL金银花供试品溶液,加水稀释至刻度,摇匀,静置30 min,测定荧光强度(F/F0),代入标准曲线方程计算出金银花供试品溶液中Fe3+的含量,并通过回收率试验考察该检测方法的准确度。
1.3 数据处理
CDs制备单因素实验数据及Fe3+检测试验数据采用Origin 2018 软件作图,正交试验及离子选择性数据分析采用SPSS 18.0软件处理。
2 结果与分析
2.1 CDs制备单因素实验
结合文献报道[31],氮掺杂CDs常采用尿素、硫脲、乙二胺、乙二胺四乙酸等含氮化合物作为表面修饰剂,通过化学反应,生成富含羧基和酰胺基等亲水性官能团的蓝色或蓝绿色荧光碳点。在CDs的合成中,首先考察了上述4种表面修饰剂对CDs合成的影响。试验数据表明,以乙二胺为表面修饰剂合成的CDs荧光最强,故后续实验均以乙二胺为表面修饰剂合成碳点。当乙二胺的加入体积为2 mL时,碳点的荧光较强,随着体积的增大反而下降,究其原因可能是因为乙二胺过量,在加热时挥发导致反应溶液温度降低,从而影响CDs的合成。
加热功率和时间均对CDs的合成有一定的影响。微波加热的功率会直接影响反应体系的温度,当功率为125 W,反应15 min时,反应混合物仍为液体。当功率调为300 W以上时,反应混合物为褐色固体,且在400 W时荧光最强。加热时间在10 min以下时,由于维持高温时间较短,合成CDs的荧光强度较低,当加热15 min时,合成CDs的荧光强度较强。同时,反应混合液中水量增加也会影响反应的温度,当微波加热功率为560 W,加水5 mL时合成CDs的荧光最强,随后呈逐渐下降趋势。
通过对单因素实验结果直观分析,当猴桃汁取样量为1 mL时,各单因素中最佳水平分别为:乙二胺加入量2 mL,加水5 mL,微波加热功率400 W,加热时间15 min。详见图1(a~d)。

图1 乙二胺用量(a)、加热时间(b)、加热功率(c)、加水量(d)对CDs荧光强度的影响Fig.1 Effects of ethylenediamine dosage(a), heating time(b),heating power(c) and water content(d) on fluorescence intensity of CDs
2.2 正交试验结果及方差分析
对正交试验数据(表3)进行方差分析,结果(见表4)表明:在制备猕猴桃碳点的条件中,微波加热功率对CDs合成有极显著性影响(P<0.01),其他因素均无显著性影响(P>0.05),其影响程度大小依次为:C>B>A>D,即微波加热功率>加热时间>乙二胺用量>加水量,结合直观分析,确定最佳的制备条件为:微波加热功率560 W,加热时间20 min,乙二胺用量2 mL,加水量5 mL。
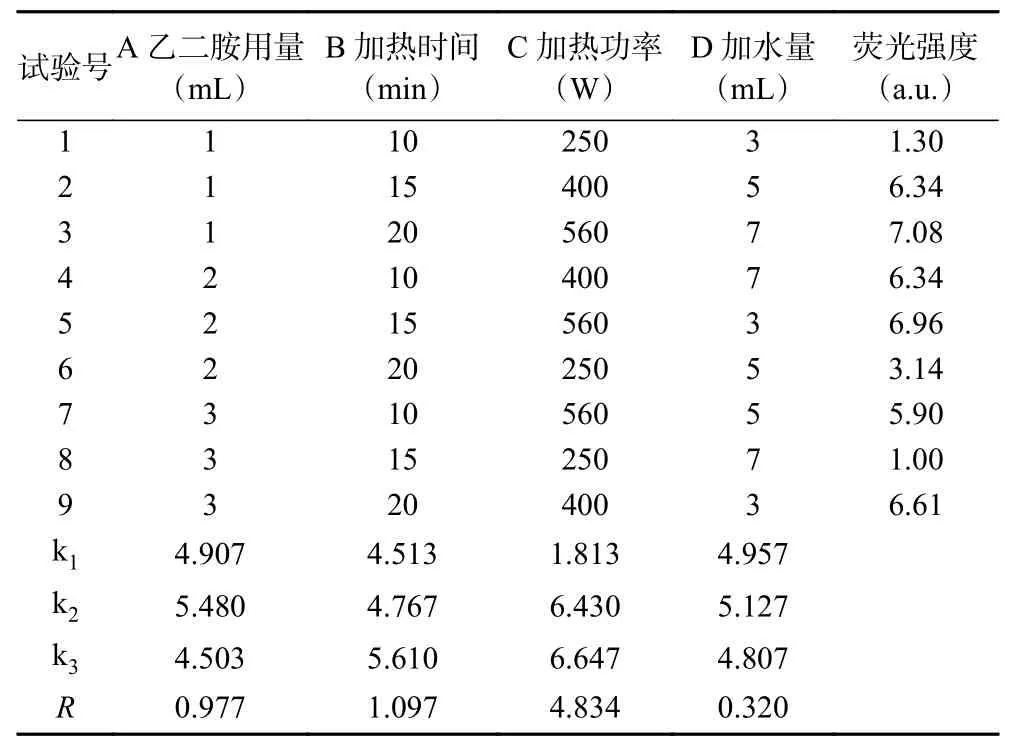
表3 正交试验结果Table 3 Results of orthogonal test

表4 方差分析结果Table 4 Analysis of variance
2.3 CDs结构表征
为了解制备的CDs组成及表面基团,对其进行透射电子显微镜及红外光谱表征,图2 (a) 为CDs的TEM,从图2 (a)中可以看出,制备的CDs呈形状均一、大小均匀、在水中分散性良好,其平均粒径约为2.5 nm。图2 (b)为CDs的FT-IR,从图2 (b)中可以看出,在3367 cm-1处的特征吸收峰为-OH或-NH-等活泼氢的伸缩振动吸收峰,2833 cm-1处的特征吸收峰为-OCH3中的C-H伸缩振动吸收峰,2946 cm-1处的特征吸收峰为C-H伸缩振动吸收峰,1030 cm-1处的吸收峰为C-O伸缩振动吸收峰。红外光谱数据表明CDs的表面含有-OH、-NH-、C-O等基团。

图2 CDs 的TEM图(a)和FT-IR光谱图 (b)Fig.2 TEM image (a) and FT-IR spectrum (b) of CDs
CDs水溶液在日光下呈微黄色,在紫外灯照射下,发射蓝色荧光。对CDs进行紫外-可见吸收光谱及荧光光谱性能测试,紫外吸收图如图3(a)所示,在约256~300 nm范围内有紫外吸收带,该吸收带主要是CDs sp2区域中π-π* 跃迁[32]。在365 nm波长紫外光的激发下,其发射峰波长为452 nm,半峰宽约为110 nm,溶液如图3(b)所示。采用硫酸奎宁(其量子产率为54.0%)作为标准参照物,为了使再吸收效应最小化,所测溶液的吸光度值均应小于0.05,测得CDs的荧光量子产率为5.6%。
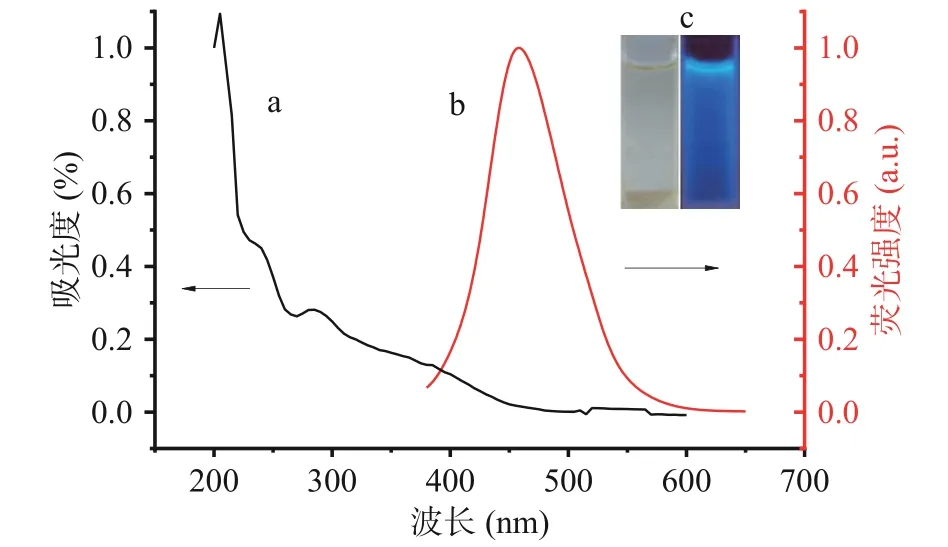
图3 CDs的紫外-可见吸收光谱图(a)和荧光发射光谱图(b)Fig.3 UV-vis absorption spectrum(a) , fluorescene emission spectrum (b)
2.4 Fe3+的检测条件的优化
如图4所示,CDs水溶液在放置5~20 min时,荧光强度呈下降趋势,20~45 min,荧光强度减弱趋缓。在pH4.2~5.5的HAc-NaAc缓冲溶液中,CDs的荧光强度随着pH的增大逐渐减弱,当HAc-NaAc缓冲溶液pH4.6时,Fe3+对CDs的荧光猝灭效果最佳。结合相关文献[33],为了排除溶液pH对CDs荧光强度的影响,故选择在pH4.6的HAc-NaAc缓冲溶液中考察Fe3+对CDs荧光强度的影响。
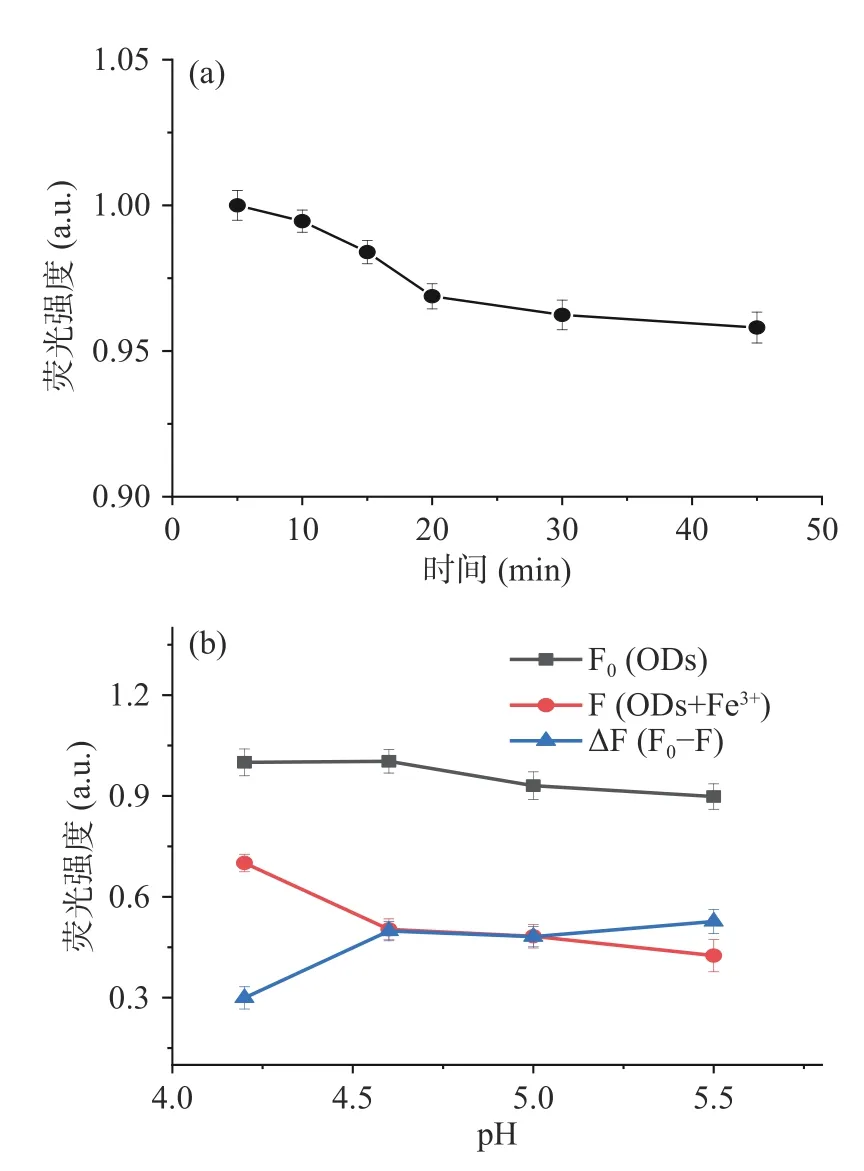
图4 放置时间影响 (a)及pH影响 (b)Fig.4 Effect of storage time(a) and pH(b)
2.5 CDs应用于Fe3+的检测
为进一步考察Fe3+对CDs荧光猝灭的动力学特征,研究Fe3+溶液浓度的变化对CDs荧光猝灭的影响,如图5(a)所示,随着Fe3+浓度的增加,CDs荧光强度逐渐减弱。当Fe3+的加入量在0.2~10 μmol/L范围内,CDs的荧光猝灭程度(F/F0)随Fe3+浓度的增加而呈线性下降,如图5(b)所示,其线性方程为Y=-0.05312c+1.01882(R2=0.9934),线性关系良好,检出限(S/N=3)为0.12 μmol/L。

图5 CDs随Fe3+浓度(0~10 μmol/L)变化的荧光光谱图(a)及CDs的荧光猝灭程度与Fe3+浓度(0.2~10 μmol/L)之间的关系曲线(b)Fig.5 Fluorescence spectra of CDs with Fe3+ (0~10 μmol/L)(a) and the curves of CDs fluorescence quenching with Fe3+(0.2~10 μmol/L)(b)
2.6 CDs对Fe3+的选择性
为研究CDs应用于Fe3+检测的选择性,在相同的浓度下,取Fe3+、Cr3+、Co2+、Ni2+、Sn2+、Zn2+、Ba2+、Fe2+、K+、Al3+、Na+、Mn2+、Mg2+、Cu2+、Ag+、Pb2+等16种常见金属离子进行试验,并进行单因素方差分析。所得试验数据均满足方差齐性要求,单因素方差分析结果表明Fe3+组与空白组及其它离子组之间差异明显,说明Fe3+对CDs 荧光猝灭作用明显,CDs应用于Fe3+的检测有较好的选择性。除Fe3+组以外的其他金属离子与空白组之间无明显差异,结果如图6所示。
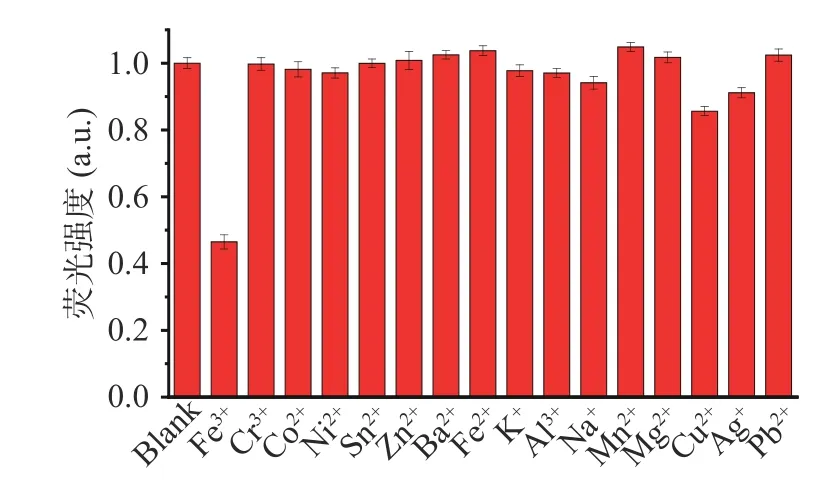
图6 CDs 对金属离子的选择性Fig.6 Selectivity of ions as using CDs probes
2.7 金银花样品中的Fe3+的检测
基于Fe3+对CDs荧光猝灭作用,试验采用荧光光度法[34]测定金银花样品中的Fe3+。为了验证该方法的准确度,试验选用常用中药材金银花为检测对象,设计加标回收试验,结果如表5。其加标回收率在98.82%~103.20%之间,相对标准偏差(RSD)介于1.67%~2.05%之间,符合中国药典[35]方法学考察要求。表6为不同方法检测金银花中Fe3+的结果。其中,本文采用的荧光光度法检出限较原子光谱法低,线性范围也相对较小,这表明该方法适合微量Fe3+的检测。
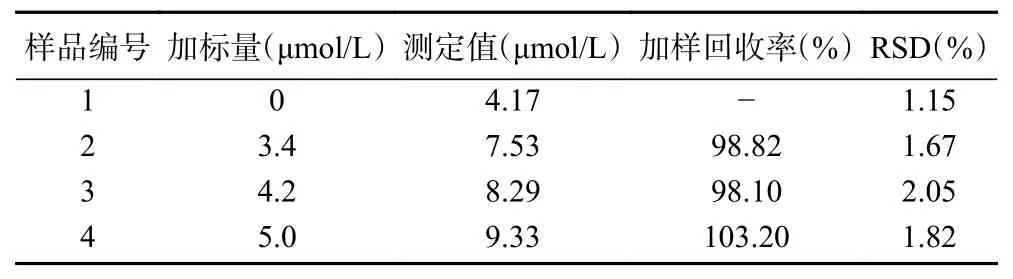
表5 金银花Fe3+的检测与加样回收率(n=3)Table 5 Determination results and recovery of Fe3+ in honeysuckle (n=3)

表6 与其他检测Fe3+的方法比较Table 6 Comparison with other methods for determination of Fe3+
3 结论
近年来,利用天然产物合成碳点,因其绿色环保、无毒无害,成为荧光探针研究的热点。本文以猕猴桃为生物质碳源,乙二胺为表面修饰剂,采用微波法一步合成了具有功能化的猕猴桃生物质碳点,通过正交试验优化了制备工艺参数,其最佳参数为微波加热功率560 W,加热时间20 min,乙二胺用量2 mL,加水量5 mL。该参数稳定可靠,较水热法[39]合成CDs时间短,操作简便。
基于CDs荧光强度随加入Fe3+浓度的增大而呈线性减弱,进而建立一种检测铁离子的方法。本文方法相对原子光谱法,检测限低,线性关系良好,符合方法学考察要求,是一种具有选择性好、检测成本低、快速灵敏的Fe3+检测方法,可用于中药材中微量铁的检测。
