基于转录组测序的高羊茅分蘖与株高相关差异表达基因分析
2022-02-10杨志民邢瑞丁鋆嘉庄黎丽
杨志民,邢瑞,丁鋆嘉,庄黎丽
(南京农业大学草业学院,江苏 南京 210095)
分蘖和株高是禾本科牧草和草坪草的两个重要表型特征。高植株多分蘖的牧草品种地上生物量大,可以获得高产;矮植株多分蘖的草坪草品种能够快速成坪,成坪后草坪质量高,耐践踏,少修剪,可节约养护成本。此外,在草坪草遭受诸如干旱、盐碱和热激等非生物胁迫后,分蘖对于其恢复再生也十分重要。植物分蘖发育过程受遗传和环境信号的调控,分蘖的发育一般分为两步——叶腋分生组织(axillary meristems,AMs)和分蘖芽的起始以及分蘖芽的伸长[1—2]。叶腋分生组织位于植物的叶腋处,在植物的整个生命周期内,AMs会生长出分枝或是分蘖。在模式植物腋芽缺失突变体的试验中,一些基因已经被证实是调控AMs发育的关键基因[3],例如,番茄(Solanum lycopersicum)的Gibberellic acid insensitive(GAI),Repressor of GA 1-3 mutant,Scarecrow(GRAS)同源转 录 因 子LATERAL SUPPRESSOR(LS)[4]、水 稻(Oryza sativa)的MONOCULM 1(MOC1)[5]和 拟 南 芥(Arabidopsis thaliana)的LATERAL SUPPRESSOR(LAS)[6]。MOC1功能缺失会导致分蘖数和生殖枝数下降[5]。miR164负调控LAS,可直接促进CUP SHAPED COTYLEDON1(CUC1)和CUC2转录因子的降解[7]。REGULATOR OF AXILLARY MERISTEMS1(RAX1)是另一种重要的AMs起始因子,可直接正向调控CUC2在边界区域的特殊表达。RAX1还可以和RAX2、RAX3协同调控AMs的起始[8]。TEOSINTE BRANCHED1,CYCLOIDEA,PCF(TCP)转录因 子家族的FINE CULM 1/PsBRANCHED1/BRANCHED 1(FC1/PsBRC1/BRC1)等基因在腋芽处特异表达,通过整合各种通路如激素响应和光形态建成等负调控植株分枝[9—11]。目前对调控腋芽起始和伸长的分子机制主要集中在模式植物,其他植物中侧枝/分蘖发育分子机制的研究还有很多欠缺。
植株高度受遗传和内源激素等多种因素调控。赤霉素(gibberellin,GA)可以打破植物休眠、促进种子萌发、茎节伸长和调节茎秆形态。在西瓜(Citrullus lanatus)中过表达ClTCP14a和ClTCP15可以提高内源GA含量,促使植株增高[12]。过表达GA 2oxs会使植物内源活性GA含量下降,使植株矮化,例如,在菠菜(Spinacia oleracea)中过表达SoGA 2ox3[13]、在拟南芥中过表达AtGA 2ox7和AtGA 2ox8[14]和在矮牵牛(Petunia hybrid a)中过表达PhGA 2ox1[15],植株均出现矮化表型。在GA信号转导通路中,GID1作为GA受体可与GA和DELLA蛋白结合形成复合体。DELLA蛋白是GA信号传导途径的阻遏因子,当GA浓度较高时,DELLA蛋白会被蛋白酶体降解失活,植物出现正常的GA响应反应[16]。吲哚乙酸(indole-3-acetic acid,IAA)是最先发现的植物激素,参与胚胎形成、器官分化、茎与根的伸长生长等发育过程,具有顶端优势效应,可抑制侧芽的生长。IAA可以和油菜素甾醇(brassinosteroids,BR)互作产生累加的生物学效应,可以促进细胞的伸长与分化[17]。并且IAA可以诱导DWARF4的表达,促进BR的生物学合成[18]。BR的一个重要功能是促进细胞的伸长与分裂进而促进植株地上部的生长发育。拟南芥BR缺失突变体[19]与水稻BR缺失突变体均呈现矮化表型,并且水稻在外源施用BR可以恢复正常表型[20]。
高羊茅(Festuca arundinacea)是应用最广泛的冷季型禾草,既可用于建植草坪,又是优良牧草。“Kentucky-31”(K31)是典型的高羊茅牧草品种,植株高大,但分蘖能力较弱;“Regenerate”是草坪型培育品种,优点是植株相对较矮且分蘖能力强。两个品种明显不同的表型是由哪些基因的差异表达造成的目前尚不清楚。高羊茅的基因组较大(约6 GB),当前已有一些表达数据标签(expressed sequence tag,EST)及基于非生物胁迫如抗热、抗重金属等研究所获得的转录组序列[21—22],但有关高羊茅分蘖和株高方面的转录组序列研究,目前尚未有报道。下一代测序技术和RNA测序可以分析转录组序列,挖掘差异表达基因(differentially expressed genes,DEGs),甚至可以检测尚无基因组参考的物种的基因序列。本研究目的在于获取更多的高羊茅转录组序列信息,并且比较“K31”和“Regenerate”在生长发育尤其是分蘖和株高发育上的分子机制差异,以便进一步挖掘调控高羊茅腋芽发育和植株高度的基因。
1 材料与方法
1.1 植物材料、培养条件及处理
本研究的试验材料为高羊茅品种“K31”和“Regenerate”,由美国罗格斯大学植物生理课题组提供。试验于2015年2月在南京农业大学草坪科学实验室进行。将两种高羊茅种子统一置于湿润的滤纸上,并在黑暗条件下低温春化。春化后,将种子置于光下萌发。取15株7 d龄的幼苗移栽到直径20 cm的塑料盆中,培养基质采用陶粒土。所有植物材料都栽培于环境可控的人工气候室内(MT 8070iE,河南旭邦),光周期设置为白天14 h/黑夜10 h,空气湿度控制在70%,光密度为300μmol·m—2·s—1,昼/夜温度为25oC/15oC,每周补充一次Hoagland营养液。
为了获得高羊茅de novo拼接转录组序列,分别采集“K 31”和“Regenerate”各类植物样本(各品种1个生物学重复),包括:植物全株(2周龄)、叶片、根、分蘖节、腋芽、叶耳和茎(3月龄)。将所有样本混合并储存在—80oC冰箱用于提取RNA。
为了获得“K 31”和“Regenerate”分蘖节中差异表达基因,在3月龄植株分蘖节部位取样(各品种3个生物学重复),将样品液氮速冻后于—80oC冰箱储存。
1.2 总RNA提取、cDNA文库构建与转录组测序
材料总RNA的提取使用RNeasy Plant Mini Kit(Qiagen,美国)试剂盒。采用NanoDrop(Nano-Drop Technologies,美国)和Agilent 2100 Bioanalyzer(Agilent Technologies,美国)检测样本纯度和浓度,采用琼脂糖凝胶电泳检测RNA完整性,以保证进行转录组测序的样品符合标准。
采用Illumina TruSeq RNA Sample Prep Kit(Illumina Inc.,美国)和Ultra RNA Library Prep Kit for Illumina构建cDNA文库,再用Beckman AMPure XP beads(Beckman Coulter,美国)纯化PCR产物。使用Agilent 2100对文库大小进行检测,保证测序文库质量。文库构建完成后用Bio-RAD CFX 96进行荧光定量PCR(qPCR)准确定量。采用HiSeq 2500结合Miseq 300测序平台(Illumina Inc.,美国)进行“Regenerate”和“K31”转录组深度测序用于拼接组装,采用HiSeq 2000测序平台对“Regenerate”和“K31”分蘖节进行差异表达基因分析。
1.3 高羊茅转录组序列组装、注释及分析
使用FASTX-Toolkit软件,将测序得到的原始数据进行过滤,去除接头和低质量reads(Q<30,length<50 bp)。然后通过Trinity软件对clean reads进行组装获得转录本序列,利用CAP3软件得到一套最终的单基因簇(unigenes)。利用BlastX软件将unigenes的序列比对到NCBI非冗余蛋白数据库(non-redundant protein database,NR)、蛋白质数据库(universal protein,Uniprot)、东京基因与基因组数据库(kyoto encyclopedia of genes and genomes,KEGG)和蛋白质直系同源数据库(cluster of orthologous groups,COG)中,得到unigenes的蛋白功能注释信息,然后利用WEGO软件进行unigenes的基因本体数据库(gene ontology,GO)功能分类统计。
1.4 DEGs筛选注释及Real-time PCR验证
取两个高羊茅品种的分蘖节部位转录本进行差异表达分析,以“K31”为对照组,“Regenerate”为处理组,应用RPKM(reads per kilobase per million reads)方法分析基因表达水平,以差异倍数(fold change,FC)>1或<—1且P值≤0.05为筛选标准,之后对获得的DEGs进行GO功能分类统计。在DEGs中挑选出与高羊茅生长发育相关的关键基因进行荧光定量PCR验证。采用2—ΔΔCT法分析基因的相对表达量,以α-Tubblin(Accession No.GT 051159)(F:ATGCTTTCGTCTTATGCCC;R:CTCTTGGTTTTGATGGTTGC)作 为 内 参 基 因,用Primer Premier Software(Version 5.0)设计定量PCR引物序列(表1),每个样本3次生物学重复,每个生物学重复做3次技术重复。
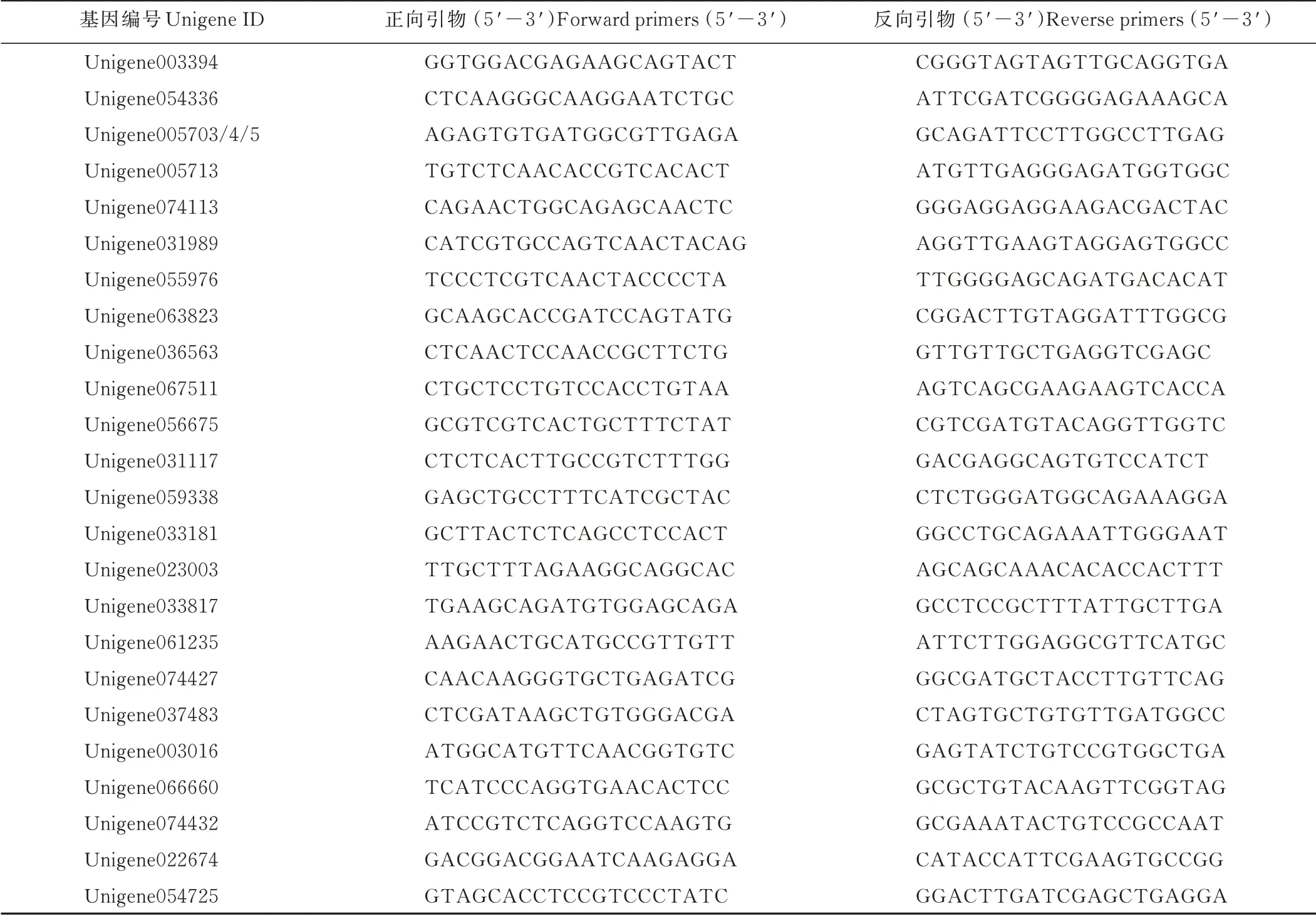
表1 PCR验证引物Table 1 PCR validation primers
1.5 植物表型观测
每个品种设计3个生物学重复,每个重复内有20株植物材料。从萌发后4周开始分别统计株高直到第7周结束,每周测量一次。将栽培基质到叶尖的最大长度定为株高。
统计种子萌发后4~7周的分蘖数和腋芽数,在体视显微镜(Olympus,日本)下观察腋芽并拍照。
1.6 数据分析
采用一般线性统计模型(SAS 9.0,美国)对生长和生理数据进行方差分析(ANOVA)。采用Fisher最小显著性检验(LSD,P<0.05)进行均数分离。
2 结果与分析
2.1 两个高羊茅品种的表型差异
本研究中,“K31”和“Regenerate”在生长发育方面有不同的表型特征(图1A,B)。从种子萌发后4周开始统计分蘖数和腋芽数、株高、茎秆直径、叶细胞大小等生长数据。萌发后第4周及第5周时,“K31”和“Regenerate”的分蘖数没有显著差异,但是在第6周和第7周时差异显著(图1C)。在株高方面,萌发后第4周,“K 31”植株高度为24.53 cm,显著高于“Regenerate”的16.1 cm。萌发后第7周,“K 31”已经长至50.78 cm,而“Regenerate”为28.01cm(图1D)。两者茎叶(此处茎指的是层层包裹的叶鞘)生长速率也有明显不同,“K31”的茎比“Regenerate”更为粗壮,萌发后第4周时“K31”茎秆直径为1.33 mm,“Regenerate”直径为1.22 mm。萌发后第6周时,“K31”的直径为2.35 mm,“Regenerate”为1.91 mm(图1E)。表皮细胞面积同样有显著差异,萌发后第4周时,“K 31”细胞面积为8887.29μm2,“Regenerate”细胞面积为6536.32μm2;第7周时,细胞面积缩小,“K31”细胞面积为6674.48μm2,“Regenerate”细胞面积为5629.03μm2(图1F)。
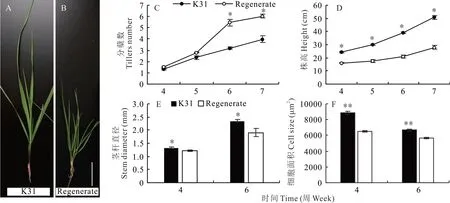
图1 “Regenerate”和“K 31”植株的表型差异比较Fig.1 Compar ison of phenotypic differ ence between“K 31”and“Regenerate”plants
2.2 组装与测试结果分析
2.2.1高羊茅测序结果和组装 “K31”和“Regenerate”两个品种高羊茅组成的RNA-seq文库通过Hiseq 2500结合Miseq 300测序平台进行测序。“K31”获得76673214条raw reads,“Regenerate”获得88475512条raw reads(表2),原始测序数据已提交到NCBI的序列片段归档(sequence read archive,SRA)数据库(Bioproject ID:PRJNA 669484)。将原始序列数据中的接头污染数据、低质量数据以及重复冗余数据去除后,对“K31”的76533900条clean reads和“Regenerate”的88345910条clean reads进行后续分析。Clean reads分别占比97.60%和97.81%(表2),证明所得到的数据是优质数据。“K31”和“Regenerate”的clean reads共获得77872条unigenes,unigenes长度范围从200 bp跨越到13402 bp,平均长度为611.086 bp(N50=920 bp,N90=262 bp)(表3)。Unigenes的GC含量为53.2%(表3),接近水稻的GC含量(55%)。

表2 de novo RNA-seq详细序列信息Table 2 Detail sequence information for de novo RNA-seq

表3 样品RNA-Seq数据汇总Table 3 Summary of sample based on the RNA-Seq data
2.2.2高羊茅转录组unigenes的物种分布与COG功能注释 利用BlastX将组装得到的77872条unigenes分别与NR、Uniprot/Swiss-Prot、KEGG、COG和GO数据库(E-value<1e—5)进行比对的结果显示:共有60552条unigenes获得蛋白功能注释,占总unigenes的77.76%。其中有8206条unigenes在所有数据库都有获得功能注释(图2,表4)。从注释匹配的物种分布来看,来自二穗短柄草(Brachypodium distachyon)的注释最多,15882条注释,占总注释的26.50%;其次为大麦(Hordeum vulgure),13594条注释,占总注释的22.68%。说明在已知基因组信息的物种中,高羊茅与二穗短柄草和大麦的序列相似度较大。其他与高羊茅序列相似度较大的物种依次为山羊草(Aegilops tauschii)(17.58%)、乌拉尔图小麦(Triticum urartu)(11.87%)、粳稻(Oryza sativaJaponica Group)(4.53%)和玉米(Zea mays)(4.50%)等。除此之外,有388条unigenes(0.65%)没有比对到任何物种(图3)。高羊茅unigenes与COG数据库对比结果表明:共有13767条unigenes被归纳到24个不同功能分类当中。其中,有3801条unigenes为一般功能预测蛋白(general function prediction only),占总功能序列的4.88%,在所有功能基因中占比例最高;然后依次是蛋白质翻译后修饰—蛋白质周转—分子伴侣(posttranslational modification,protein turnover,chaperones,1970 unigenes,2.53%)、转录(transcription,1766 unigenes,2.27%)和信号转导机制(signal transduction mechanisms,1628 unigenes,2.09%)。只有少数序列归纳到细胞能动 性(cell motility,43 unigenes,0.06%)和 核 结 构(nuclear structure,6 unigenes,0.01%)(图4)。
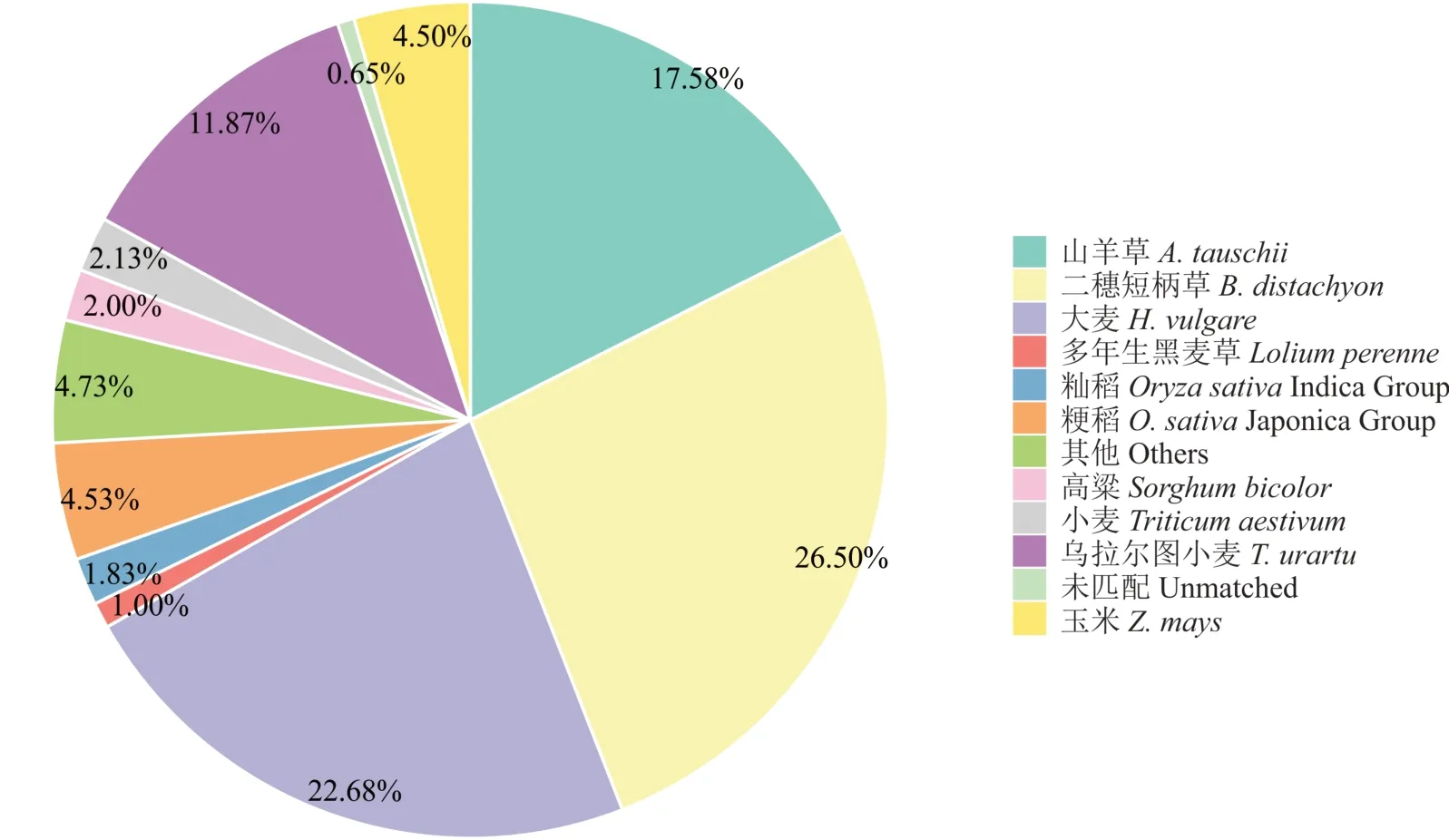
图3 物种分布结果非冗余注释Fig.3 Species distr ibution of the results of non-r edundant(NR)annotation
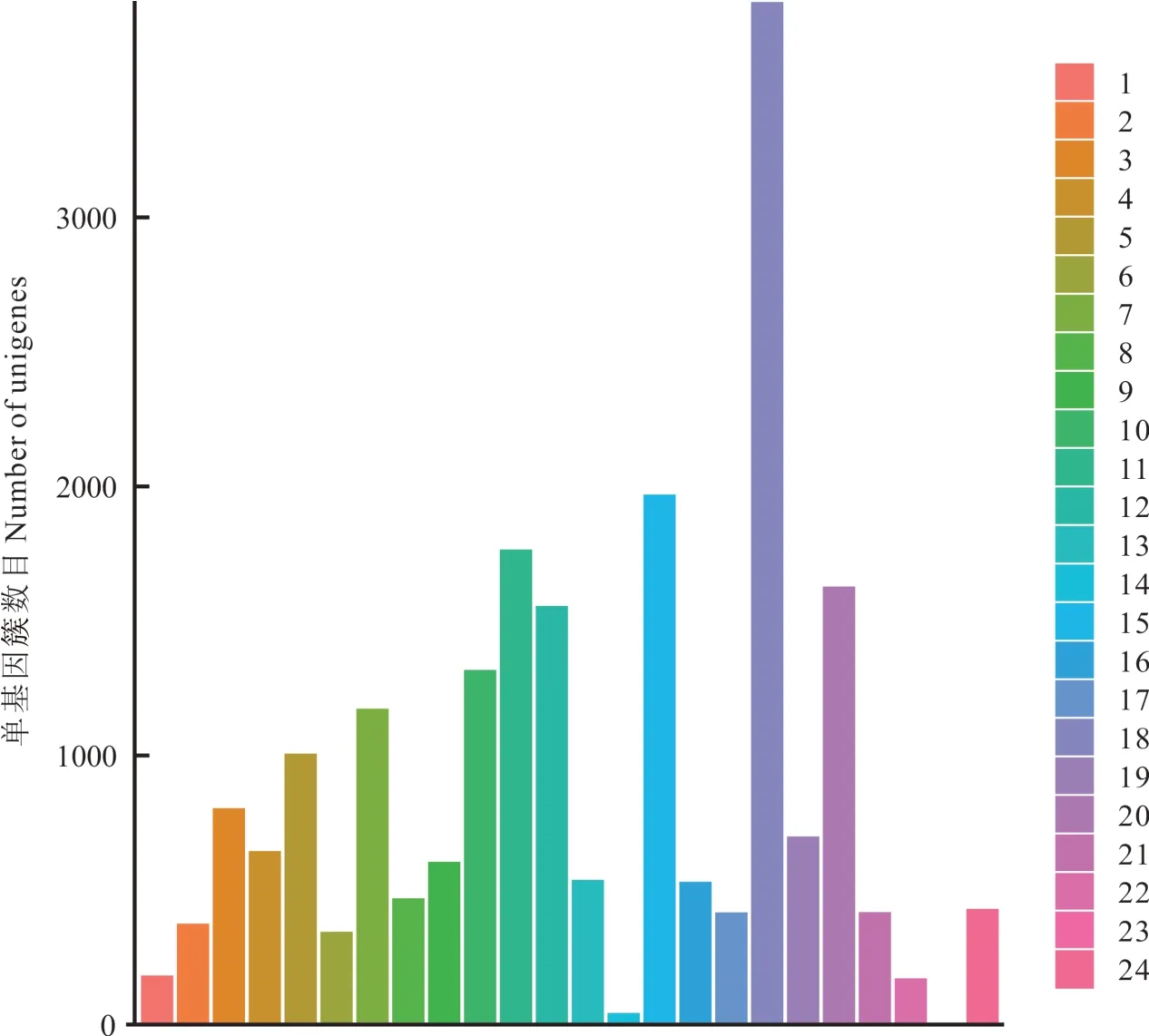
图4 高羊茅unigenes COG注释分类Fig.4 Cluster of orthologous group(COG)classif ication of unigenes in F.arundinacea
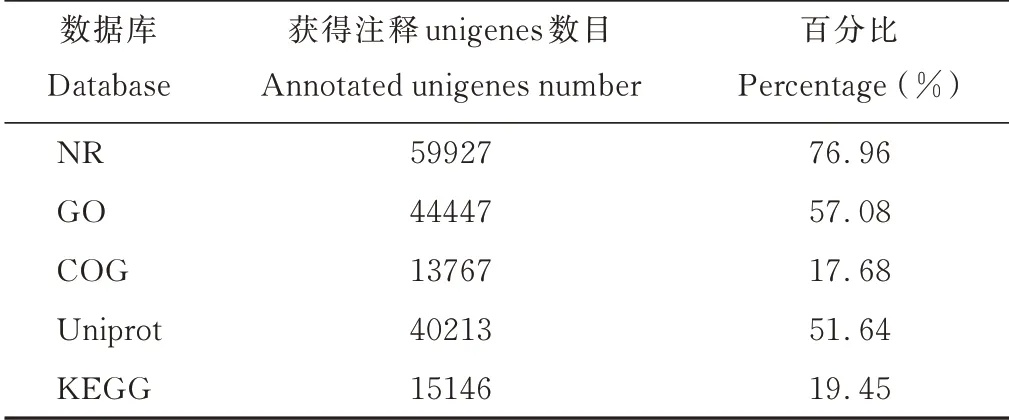
表4 数据库BLAST分析结果Table 4 BLAST analysis results against important public databases
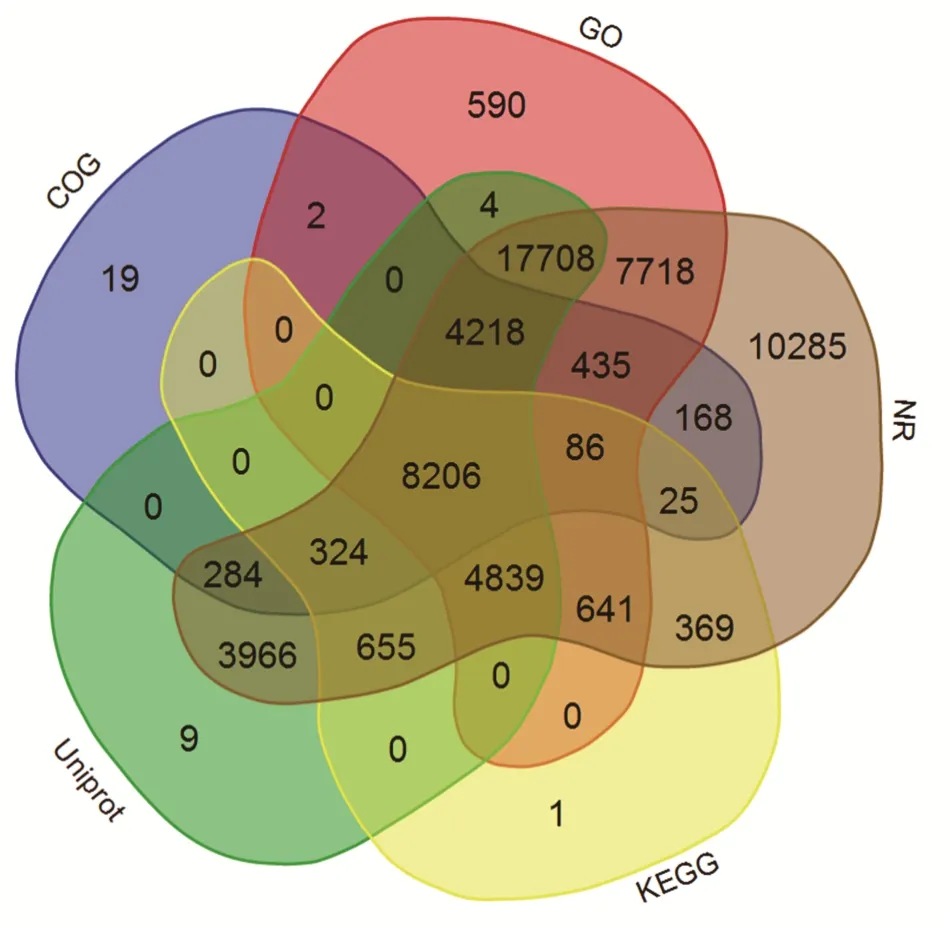
图2 获得注释的unigenes在5个数据库的分布Fig.2 Distribution of unigenes annotated in five databases
2.2.3高羊茅转录组unigenes的GO注释及分类高羊茅转录组unigenes的GO功能注释所分析的结果显示:共有44447条unigenes(57.08%)获得功能注释,分布在48个亚类中。其中,有28316、37017和31216个unigenes分别注释到细胞组分、分子功能和生物过程(图5)。在细胞组分的12个功能亚类中,其中以细胞(cell)和细胞组分(cell part)较多,二者均占该类别的36.1%。其次为细胞器(organelle)和大分子复合物(macromolecular complex),分别占该类别的25.6%和7.0%。在分子功能所包括的13个亚类中,捆绑(binding)和催化活性(catalytic activity)占比最多,分别占比33.3%和26.8%。在生物过程的23个功能亚类中,代谢过程(metabolic process)和细胞过程(cellular process)占比较多,分别为31.0%和30.4%。总的来看,大量unigenes分布在细胞、细胞组分、细胞过程和代谢过程等亚类中。
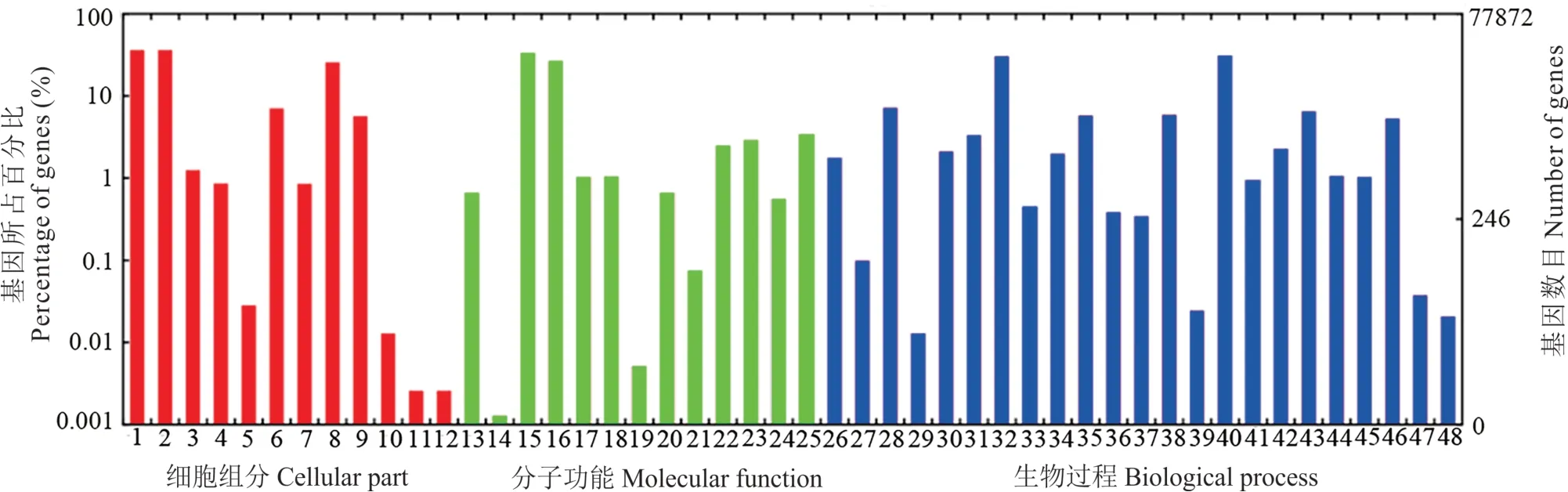
图5 高羊茅unigenes GO注释分类Fig.5 Gene ontology(GO)classification of unigenes in F.arundinacea
2.2.4高羊茅转录组KEGG代谢通路分析 通过对比KEGG数据库,高羊茅转录组测序结果显示:共有15146条unigenes获得了注释,占总数的19.45%,分布在31个代谢通路上(图6)。按照获得注释的unigenes数目进行排序,翻译(translation,1648 unigenes,10.88%)所获注释最多,然后依次是信号转导(signal transduction,1410 unigenes,9.31%)、碳水化合物代谢(carbohydrate metabolism,1404 unigenes,9.27%)、折叠,分类和降解(folding,sorting and degradation,1350 unigenes,8.91%)、氨基酸代谢(amino acid metabolism,1103 unigenes,7.28%)、运输和分解代谢(transport and catabolism,917 unigenes,6.05%)和内分泌系统(endocrine system,861unigenes,5.68%)等代谢通路。感觉系统(sensory system,35 unigenes,0.23%)和信号分子与互作(signaling molecules and interaction,12 unigenes,0.08%)所获注释较少。获得注释的代谢通路几乎代表了高羊茅发育的所有代谢过程,为进一步开展高羊茅的功能基因组学研究提供了丰富的数据资源。
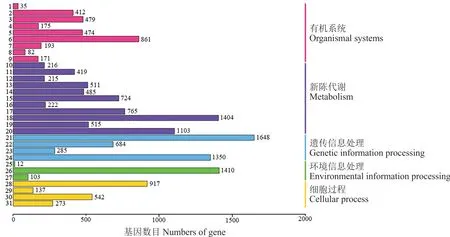
图6 高羊茅unigenes KEGG注释分类Fig.6 KEGG classif ication of unigenes in F.arundinacea
2.3 高羊茅差异表达转录本分析
2.3.1高羊茅DEGs的数目分析 基于以上的高羊茅转录组测序获得的从头拼接组装序列,我们分别对“K31”和“Regenerate”的分蘖节部位进行RNA-Seq分析,以期揭示调控两个高羊茅品种生长发育的DEGs。以差异倍数>1或<—1且P值≤0.05为筛选标准,共获得3014条DEGs。其中“Regenerate”相较于“K31”有1573条基因上调,1441条基因下调。
2.3.2差异基因GO分类与功能富集 对两个高羊茅品种的DEGs进行GO富集分析结果显示:3014条DEGs的功能注释被分为生物过程、分子功能和细胞组分三大类。三大功能分类又可进一步划分为68个功能亚类。其中生物过程包括34个功能亚类;细胞组分包括16个功能亚类;分子功能最少,只有18个功能亚类(图7仅显示三大类中富集最显著的10个亚类)。在生物过程所涵盖的34个亚类中,以细胞组分生物发生(cellular component biogenesis)、细胞大分子复合物亚基组织(cellular macromolecular complex subunit organization)、DNA包装(DNA packaging)、染 色 体 组 织(chromosome organization)和 染 色 质 组 装 或 拆 卸(chromatin assembly or disassembly)等10个亚类最为富集。细胞组分的16个功能亚类中,细胞内非膜细胞器(intracellular non-membranebounded organelle)、蛋白质-DNA复合物(protein-DNA complex)、核小体(nucleosome)、染色体组分(chromosomal part)和染色质(chromatin)等7个亚类最为富 集,其 次是细胞组 分(cell part)、细 胞(cell)和大 分子复合物(macromolecular complex)3个亚类。分子功能的18个功能亚类中,以肽链内切酶抑制剂活性(endopeptidase inhibitor activity)、酶抑制剂活性(enzyme inhibitor activity)、天冬酰胺酶活性(asparaginase activity)等10个亚类最为富集(图7)。综合分析,高羊茅差异基因的GO功能富集,以生物过程相关基因的差异性最为明显。
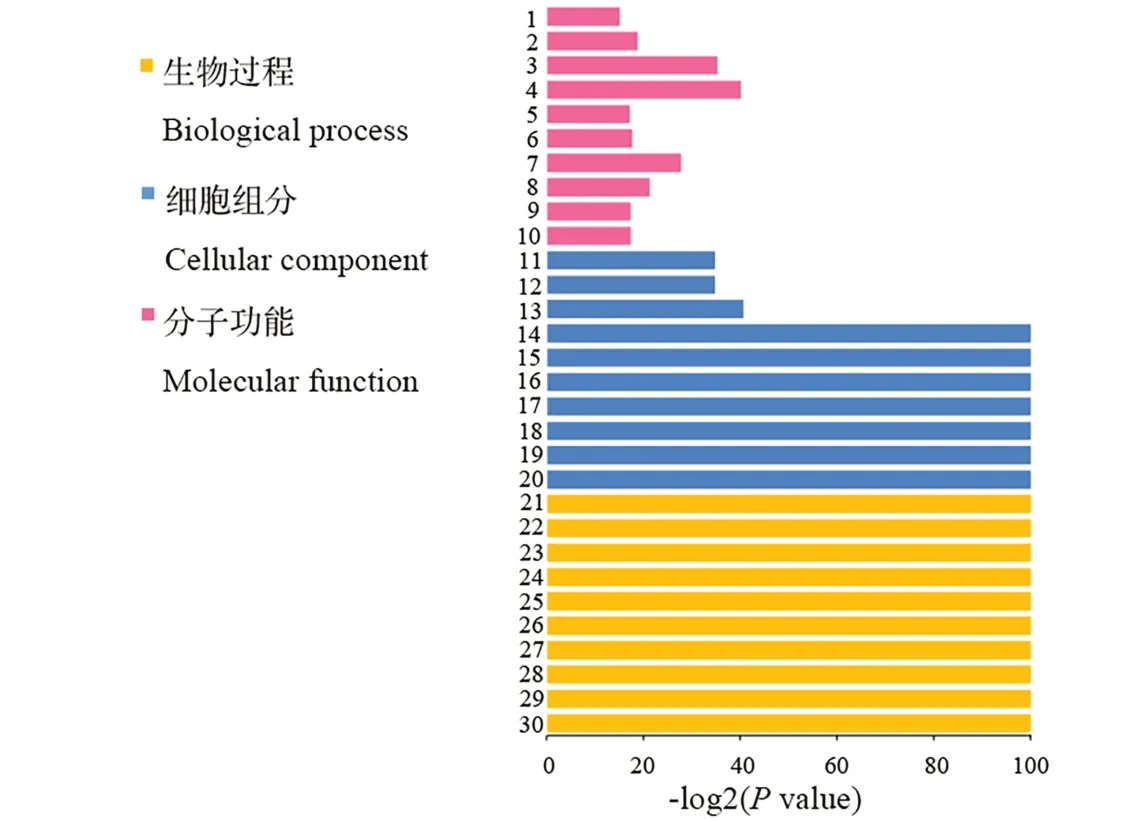
图7 高羊茅DEGs的GO功能分析Fig.7 Functional categorization of assembled unigenes based on gene ontology(GO)classification in crowns of F.arundinacea
2.4 关键通路基因转录表达分析
2.4.1转录因子 转录因子作为能调控基因表达功能的蛋白质分子,在植物的生长发育过程中以及应对环境胁迫时都起到了重要的作用。根据BLAST数据分析,许多蛋白可能是来自不同家族的转录因子。在所有DEGs中,发现有42个转录因子出现差异表达,这些转录因子来自不同的家族,包括MADS、TCP、ARF、AP2/ERF和bHLH等多种类型(表5)。MADS-box家族基因功能主要与细胞分化及其相关发育过程有关。有6个MADS转录因子存在差异表达,其中有5个转录因子(Unigene003445;Unigene008312;Unigene032888;Unigene067434;Unigene049455)上调,1个转录因子(Unigene016122)下调。TCP是植物特有的转录因子,研究中发现了5个TCP转录因子(Unigene032517;Unigene063740;Unigene010687;Unigene022674;Unigene010684)存在差异表达,且全部下调。3个ARF转录因子存在差异表达,其中有2个转录因子(Unigene027288;Unigene003404)下调,1个转录因子(Unigene003016)上调。AP2/ERF转录因子是植物特有的一类转录因子家族,1个AP2转录因子(Unigene038874)下调,6个ERF转录因子存在差异表达,其中5个转录因子(Unigene077012;Unigene066363;Unigene000226;Unigene059623;Unigene051413)上调,1个转录因子(Unigene016424)下调。植物bHLH转录因子参与调解各种信号转导及合成代谢途径,共有3个bHLH转录因子呈现差异表达,2个转录因子(Unigene067511;Unigene067617)上调,1个转录因子(Unigene017047)下调。
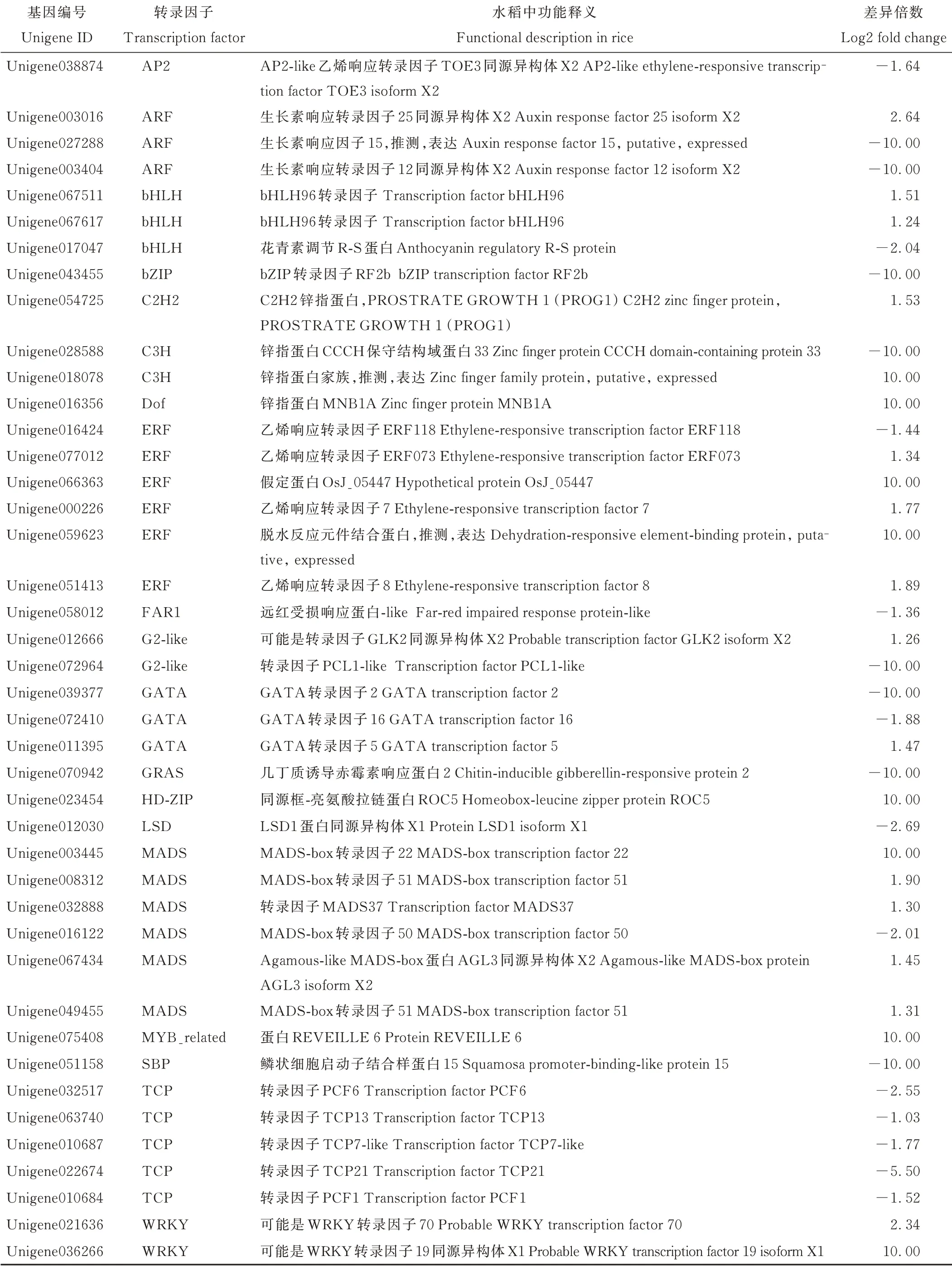
表5 差异表达转录因子Table 5 Differentially expressed transcription factors
2.4.2激素代谢及信号转导 本研究中,有85条unigenes与脱落酸(abscisic acid,ABA)、生长素、乙烯(ethylene,ETH)、细胞分裂素(cytokinin,CTK)、赤霉素、茉莉酸(jasmonic acid,JA)、油菜素甾醇和水杨酸(salicylic acid,SA)等8种植物激素相关,并且在“Regenerate”和“K31”之间存在差异表达。其中与ABA、IAA和ETH相关的unigenes条数较多。还有8条unigenes参与植物激素互作,IAA-ETH(Unigene003404)、ABA-ETH(Unigene066363、Unigene000226)、ETH-JA(Unigene023000)、ABA-GA(Unigene012266)、ABA-JA(Unigene 046376)、IAA-BR-CTK(Unigene016424)和IAA-ETH-JA(Unigene058662)。在ABA相关的DEGs中有18个基因上调,8个基因下调。下调基因中有3个醛氧化酶基因(Unigene072636、Unigene030806、Unigene 074427)。上调基因中有2个富含半胱氨酸的受体样蛋白激酶(Unigene060943、Unigene034794)、2个AP2/ERF转录因子(Unigene066363、Unigene000226)和1个转录抑制因子(Unigene000154)。与IAA相关的DEGs中有10个上调基因,9个下调基因。其中有1个生长素原初响应基因(Aux/IAA)(Unigene046724)和1个核糖体蛋白基因(Unigene046971)下调;上调基因中有1个糖基水解酶基因(Unigene074290)和1个dof蛋白(Unigene016356)。3个生长素应答因子中2个基因下调(Unigene003404、Unigene027288),1个基因上调(Unigene003016)。与ETH相关的DEGs有12个上调基因,9个下调基因。其中有3个脱氢酶基因(Unigene003249、Unigene071207、Unigene071208)和1个ACC氧化酶基因(Unigene064634)上调;2个剪接因子基因(Unigene004991、Unigene 004995)和1个热激蛋白基因(Unigene037362)下调。与GA相关的6个DEGs全部上调表达,其中有3个扩张素前体(Unigene037534、Unigene031989、Unigene074120)和1个开花促进因子(Unigene026443)。之外还有7个DEGs与JA相关(3个基因上调,4个基因下调);3个DEGs与CTK相关(2个上调,1个下调);2个DEGs与SA相关(1个上调,1个下调);1个DEGs与BR相关(1个下调)(表6)。

表6 植物激素相关差异表达基因Table 6 Plants hormone related DEGs

续表Continued Table
2.5 DEGs的荧光定量PCR验证
为了验证通过RPKM值统计比较得到的DEGs数据,挑选5种类型(转录因子、激素相关基因、扩张素基因、致病相关蛋白和未知功能基因),共24个DEGs进行实时定量PCR(real-time quantitative PCR,qRT—PCR)验证。选择FaACTIN4(ACTIN-related protein 4)作为荧光定量PCR验证的内参基因。qRT—PCR检测到的所有基因的表达水平与RNA-Seq数据具有较强的一致性(图8),表明这些RNA-Seq数据准确有效,可用于高羊茅的基因表达图谱分析。
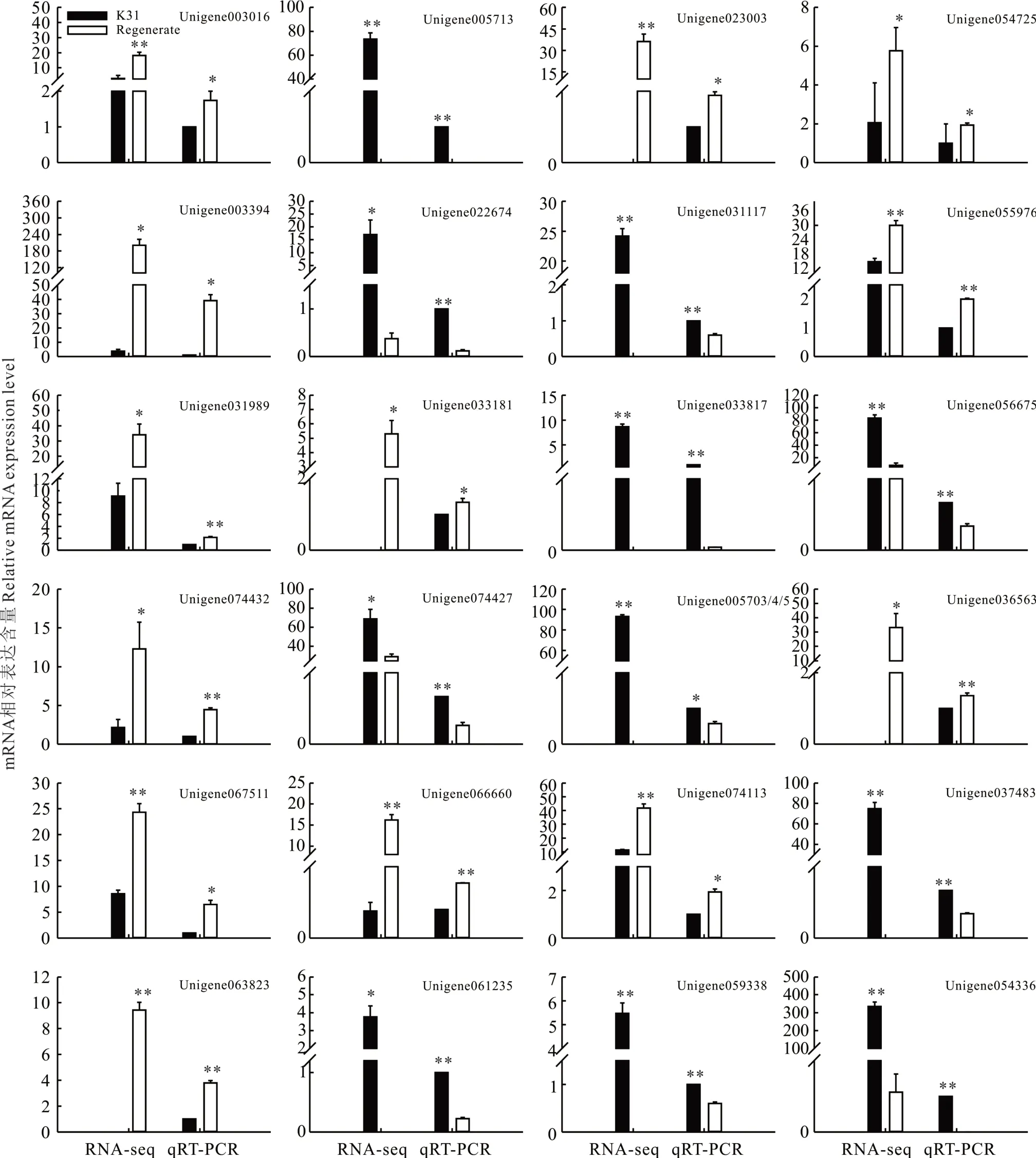
图8 DEGs的荧光定量PCR验证Fig.8 Validation of RNA-seq by q RT-PCR
3 讨论
Illumina Hiseq高通量转录组测序分析结果显示:共获得77872条unigenes,与NR、Uniprot、COG、GO和KEGG等数据库进行比对,共60552条unigenes获得功能注释。此外从“Regenerate”和“K31”两个高羊茅品种的分蘖节部位筛选出3014个DEGs,“Regenerate”相较于“K31”,上调基因数为1573个,下调基因数为1441个。这些上调和下调的基因对于揭示高羊茅参与株高和分蘖等形态发育的调控基因及其相关代谢途径具有重要作用。通过GO富集分析,明确了DEGs的基因功能以及参与调控的信号通路。
本研究中发现42个存在差异表达的转录因子,包括MADS、TCP、MYB、WRKY、bHLH等19种类型。其中,MADS家族基因广泛存在于植物、动物和真菌中,它们在生长发育和信号转导中起着重要作用。在植物中,它们几乎参与到植物发育的各个方面,最主要体现在调控开花时间和花器官形成方面,以及根和种子的发育。著名的ABCDE模型解释了在花的不同部位,不同转录因子对花器官形成的作用,其中许多转录因子来自MADS家族[23]。此外,前人研究发现OsMADS57可以影响植物分蘖/分枝的形成,它可以调节D14(独脚金内酯受体),并与OsTB1相互作用,共同调控水稻分蘖[24]。MADS转录因子还可以通过调控植物体内激素水平影响植物发育,OsMADS22负调控BR的合成[25],而BR在植物生长发育过程中发挥重要的作用,在植物的不同器官均含有BR[26],它可以调控细胞伸长和分裂、光形态建成和维管束发育等过程[27]。BR受体过表达拟南芥出现叶片表皮和叶肉细胞体积增大现象[28],本研究中“Regenerate”的表皮细胞体积小于“K 31”,且BR相关差异基因(Unigene016424)呈下调模式,这可能与BR对叶片细胞体积的调控相关。关于MADS参与草类植物生长发育的报道相对较少,本研究所鉴定的6个MADS-box unigenes是否影响了两种高羊茅在表型上的差异还有待进一步研究。
TCP是植物特有的转录因子,以玉米的TEOSINTE BRANCHED 1(TB1)、金鱼草(Antirrhinum majus)的CYCLOIDEA(CYC)和水稻的PROLIFERATING CELL FACTORS1/2(PCF 1、PCF 2)的首字母命名。TCP在调控植物细胞周期及根、茎、叶、花、种子等器官发育过程中发挥着重要的作用[29]。TCP家族通常在快速生长的组织器官中表达较多,说明这种转录因子与植物细胞的分裂分化与组织器官的发育有密切关系。在拟南芥中,AtTCP20能够与CYCB1的基因元件相结合,进而调控细胞的分裂与生长[30]。AtTCP18是玉米TB1的同源基因,能够抑制腋芽的发生,AtTCP18缺失突变体呈现大量侧枝的表型,并且参与生长素调控侧枝发育的信号网络[9]。AtTCP14和AtTCP15参与细胞分裂素的信号转导途径,并且AtTCP14和AtTCP15双突变体植株出现矮化性状,这与GA-DELLA的信号转导途径相关[31—32]。从我们的RNA-Seq数据可知,TCP转录因子的表达量全部下调,结合“Regenerate”相较于“K 31”分蘖更多,植株更矮的表型,TCP转录因子的差异表达可能是两种高羊茅出现表型差异的原因之一。
WRKY是高等植物中第七大转录因子家族,普遍参与植物的生长发育以及胁迫响应机制。WRKY转录因子家族每一个成员的功能不尽相同,在植物生长发育方面的影响相对复杂,很多研究发现WRKY转录因子既可以调控开花时间、促进植株发育也可以诱导植株矮化。在前人的研究中,大豆(Glycine soja)GsWRKY 20能够控制植物的开花时间,过表达GsWRKY 20植株比野生型开花更早[33]。OsWRKY 53缺失突变体出现叶倾角变小和植株变矮等生长受阻性状[34],但过表达OsWRKY 78可促进水稻茎秆伸长[35]。拟南芥中过表达OsWRKY 72植物出现侧枝生长增强的现象[36]。在拟南芥中发现AtWRKY 71能够促进开花并且调节植株分枝发育[37],过表达AtWRKY 15可促使叶片扩增[38]。在棉花(Gossypium hirsutum)中过表达GhWRKY 15会出现茎秆伸长的性状[39]。但是,过表达OsWRKY 11水稻出现了植株矮化的表型,这一过程主要受CTK、IAA、GA和BR的调控[40]。过表达OsWRKY 13[41]和OsWRKY 89[42]使水稻生长发育迟缓,植株变矮。在我们的研究中共发现2个差异表达WRKY转录因子,均呈现上调的表达模式,结合“K 31”和“Regenerate”的性状表现,在株高和分蘖上的差异可能与WRKY转录因子相关,但是由于WRKY转录因子家族成员众多,且在不同的物种中起到的作用也不尽相同,WRKY在高羊茅生长发育过程起到的作用还需进一步挖掘。
对多种物种的研究表明,芽中ABA的含量与芽活力呈负相关[43]。外源ABA抑制了豌豆(Pisum sativum)[44]、拟南芥[45]和番茄芽的生长[46],ABA生物合成抑制剂促进了月季(Rosa hybrida)芽的生长[47]。有研究认为,ABA与玉米素核苷(trans-zeatin-riboside,ZR)可协同调控分蘖发育,ZR/ABA低时,玉米分蘖发育受到抑制,ZR/ABA高时,促进玉米分蘖的发育[48]。IAA和CTK共同调控植物分蘖芽的休眠与发育,两者的调控作用相反。IAA的极性运输被认为是抑制分枝发育的主要因素,可以抑制芽的起始[49]和伸长[50]。CTK可以解除顶端优势,诱导花芽分化,促进分蘖芽的萌发。Aux/IAA是生长素原初响应基因,可以对生长素做出快速响应。在水稻中过表达OsIAA 1导致水稻对生长素敏感性减弱,出现植株矮化,侧根增多的现象[51]。在拟南芥中过表达杨树(Populus)Ptr IAA 14.1基因,植株呈现矮化和侧枝增多的表型[52]。前人的多项研究表明ABA和IAA相关基因可以参与调控植株高度和分蘖发育,在研究中注释到的ABA相关DEGs主要参与ABA代谢和信号转导,IAA相关DEGs不参与代谢,主要参与IAA的信号转导。因此可以在之后的研究中从激素代谢和信号转导的角度进一步探讨具体是哪些DEGs直接参与影响“K31”和“Regenerate”之间的表型差异。
GA可以促进种子萌发、茎秆伸长、叶片伸展和抑制分蘖芽的发育。前人研究发现,在水稻中过表达OsGA 2ox1[53]、OsGA 2ox5[54]和OsGA 2ox6[55]会使植物体内活性GA含量下降,出现植株变矮、分蘖增多的性状。外源喷施GA还可以有效控制植物分蘖的发生[56]。本研究共得到6个与GA相关的DEGs,全部呈上调表达模式。其中有3个是扩张素前体,1个是糖基水解酶。扩张素蛋白能够改变细胞壁结构,使细胞壁松弛,促进细胞伸展与分裂;糖基水解酶在植物中主要起到调控生长发育和响应环境胁迫的功能[57]。这些基因间的差异表达可能是“K 31”和“Regenerate”表型差异的部分原因。GA的信号转导通路与GRAS家族的转录因子密切相关,GRAS家族转录因子参与了植物侧枝形成、光信号转导、茎秆发育和赤霉素信号转导等多种生理生化过程。DELLA蛋白是GRAS蛋白家族中的亚家族,参与GA与其他多种激素的信号转导,在调控植物生长发育以及应对环境胁迫中具有重要意义。DELLA蛋白可以负向调控GA的信号转导抑制植物的生长,而GA通过降解DELLA蛋白起到促进植物生长的作用[16]。本研究注释到1个GRAS家族转录因子,在水稻的研究中发现OsCIGR(chitin-inducible gibberellin-responsive)在高植株中的表达量高于矮植株[58]。该转录因子在“Regenerate”较“K31”的差异分析中呈下调表达模式,这与“Regenerate”和“K31”的株高性状一致。所以可以推测CIGR转录因子可能是引起“Regenerate”和“K31”株高产生差异的原因之一。
4 结论
本研究通过Illumina Hiseq高通量测序技术构建了不同高羊茅品种的转录组文库,初步获得了“K 31”和“Regenerate”的差异基因,挖掘到MADS、TCP和WRKY等差异表达转录因子以及众多与ABA、IAA和GA等激素代谢和信号转导相关的DEGs。为探究高羊茅生长发育的分子机制提供了基础数据和理论支持,后续可对还未明确功能和转导通路的候选基因进行进一步的功能验证。
