一种适于PCR的植物内生真菌DNA提取方法及应用
2022-02-08李瑞平宋莹莹李丽莉孙康文门兴元尹淑艳
李瑞平宋莹莹李丽莉孙康文门兴元尹淑艳
(1.山东农业大学植物保护学院,山东 泰安 271018;2.山东省农业科学院植物保护研究所,山东 济南 250100)
植物内生真菌(plant endophytes)是指其生活史的一定阶段或全部阶段生活于健康植物组织和器官内部,但不使其宿主植物表现出明显感染症状的一类真菌[1,2]。它们资源丰富、分布广泛,自然界中几乎每种植物都含有内生真菌,仅近十几年就分离鉴定了超过5 000个种,隶属于171个属[2-4]。目前鉴定内生真菌的主要方法为形态学和ITS(internal transcribed spacer)间隔测序[1,4]。
ITS核苷酸序列分析的基础为内生真菌DNA提取。传统的DNA提取方法主要包含CTAB法[4-12]、SDS法[9,12-20]、尿 素 法[21,22]、氯 化 苄法[22-27]、Chelex-100法[23,28-31]等,上述方法提取的模板质量虽然很高,但操作步骤复杂、效率低下、价格昂贵,有些还涉及到大量有机溶剂的使用,增加了对试验人员的健康危害和环境污染。ITS间隔测序仅需要进行一次简单的约600 bp的PCR扩增,对模板的质量要求极低。因此,有必要开发一种快速、简单、高效、安全的DNA提取方法,用于满足内生真菌大规模分子检测的需要。
碱裂解法最早用于大肠杆菌质粒DNA的提取,随后由Wang等[32]成功用于植物DNA的快速提取。然而该方法在真菌DNA的快速提取中鲜见报道,仅限丝核菌属(Rhizoctonia)和虫草属(Cordyceps)[33,34]。由于植物内生真菌资源丰富,且不同种类间存在细胞壁成分以及次生代谢物的差异,因此有必要评估碱裂解法在不同种属真菌的应用效果。本研究在碱裂解法的基础上,利用组织破碎仪快速研磨后高温处理,筛选出合适的NaOH浓度,评估了样本的存储条件,进而优化了DNA提取流程,并成功将其应用于68个属内生真菌快速鉴定。本试验证实了碱裂解法能够用于植物内生真菌PCR模板的快速制备,为进一步拓展该方法在整个真菌界的应用提供了新思路。
1 材料与方法
1.1 供试菌株
供试菌株共计773株,其中白僵菌为本实验室保存菌株,用于提取条件优化测试;其余772株为2021年7月在山东省130个县采集的健康玉米、大豆、棉花等植株叶片采用表面消毒培养法分离[35]、挑取尖端菌丝纯化获得[36]。
1.2 试剂与引物
NaOH、Tris等生化试剂购置于生工生物工程(上海)股份有限公司,研磨珠(3 mm)购置于济南博拓斯生物科技有限公司,PCR试剂(AG12206)购置于艾科瑞生物工程有限公司。NaOH溶液先配制成1.000 mol/L母液,随后用无菌水分别稀释为0.500、0.250、0.125、0.063、0.031、0.016、0.008 mol/L(编号为N1~N8)。Tris50为0.05 mol/L的Tris·HCl(pH 8.0),简写为T50,其他试剂均为分析纯常用试剂。引物ITS1、ITS4、LR0R、LR3R、LR7、LR5序列来源于Vilgalys Lab(https://sites.duke.edu/vilgalyslab/rdna_primers_for_fungi/,表1),由北京擎科生物科技有限公司青岛分公司合成。

表1 扩增ITS间隔和细胞核编码的核糖体大亚基rDNA(nLSU)引物序列
1.3 提取方法
(1)1.5 mL离心管中放入两个3 mm研磨珠,随后加入提取缓冲液NaOH 100 μL;(2)超净工作台中用牙签挑取少量菌丝(或菌苔、或菌块)于上述离心管中;(3)采用RETSCH MM400组织研磨仪于25 Hz破碎2 min,瞬时离心;(4)置于90℃金属浴中3~5 min(时间过长需排气),瞬时离心;(5)加入300 μL的T50,即为制备的PCR模板,-20℃或室温保存备用。
1.4 PCR反应
PCR反应体系(25 μL):ApexHF HS DNA Polymerase FS Master Mix(AG12206)12.5 μL,引物(0.3 μmol/L)0.75 μL,模板1 μL,用ddH2O补足至25 μL。反应在BioRAD MyCycler型PCR仪上进行,ITS扩增程序:94℃2 min;进行12个循环的Touchdown PCR,条件为98℃10 s,60℃(-0.5℃/Cycler)10 s,72℃10 s;98℃10 s,54.5℃10 s,72℃10 s,共34个循环;最后72℃2 min,10℃保存。nLSU(LROR/LR3R/LR5/LR7)扩增程序:94℃2 min;98℃10 s,49℃10 s,72℃30 s,共34个循环;72℃2 min,10℃保存。
1.5 检测方法
采用1.2% TAE琼脂糖电泳(6 V/cm,30 min)进行检测,EB染色,UVP凝胶成像系统拍照。PCR产物委托青岛蔚来生物科技有限公司进行Sanger测序。
2 结果与分析
2.1 不同缓冲液制备DNA溶液的状态
以白僵菌为试材,采用11种提取缓冲液按照1.3进行DNA提取,其中样本溶液状态、固体悬浮物量以及悬浮物状态观察点为1.3中第(3)步骤瞬时离心前,而离心后沉淀状态为第(4)步瞬时离心后。结果(表2)表明,N1和N2两种缓冲液裂解菌体后呈现均一状态,其他缓冲液裂解不彻底,特别是N5、N6、N7、N8、T50、TE、DDW裂解菌体后存在大量组织碎片。因此,N1和N2两种缓冲液裂解菌体效果最好,本着节约成本考虑,后续测试选择N2缓冲液。
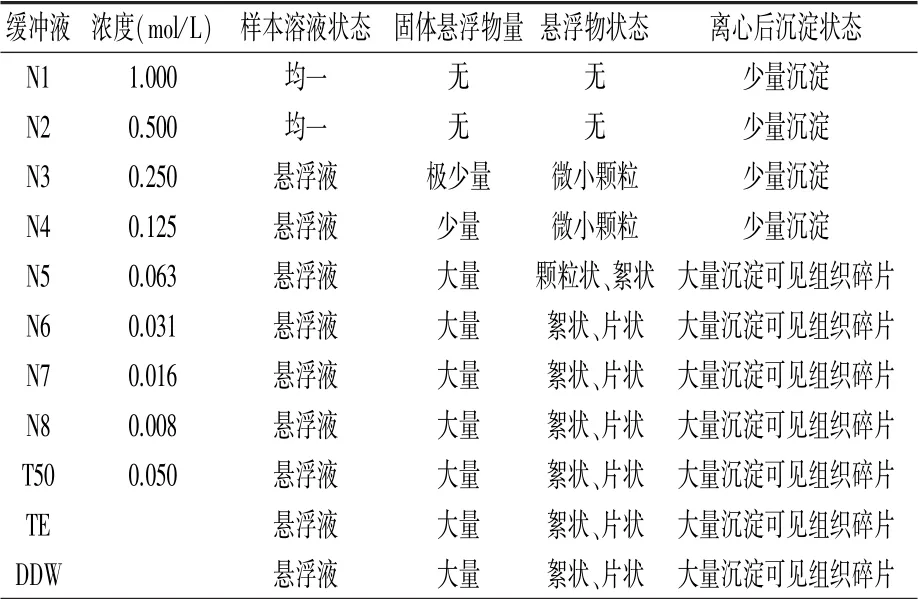
表2 不同缓冲液制备DNA模板样本的状态
2.2 NaOH浓度对PCR扩增的影响
为了探索NaOH浓度对于PCR扩增的抑制程度,进而明确碱裂解样本的稀释倍数,不同浓度NaOH制备的DNA样本不使用T50稀释,直接进行ITS扩增。由图1可知,0.250 mol/L(N3)及以上(N1、N2)的NaOH浓度可完全抑制PCR反应,0.125~0.008 mol/L(N4-N8)的NaOH对PCR扩增无影响。结合前述样本的裂解程度及溶液状态,选择0.500 mol/L NaOH制备的样本T50稀释4倍(0.125 mol/L NaOH)作为PCR模板。

图1 NaOH浓度对PCR扩增效果的抑制作用检测
2.3 不同缓冲液制备模板对PCR扩增效果的影响
按照1.3方法制备样本,其中T50、TE、DDW粗提样本不稀释直接使用,并取少量菌丝体直接PCR。由图2可知,0.500~0.008 mol/L(N2~N8)的NaOH经T50稀释后以及T50、TE、DDW粗提样本均可产生有效扩增;1.000 mol/L(N1)的NaOH制备样本无扩增产物;菌丝体直接PCR扩增条带极其微弱。该结果进一步表明0.500 mol/L的NaOH粗提样本稀释后,可用于真菌快速PCR鉴定。

图2 ITS1/ITS4引物对11种缓冲液制备样本及菌丝体的PCR检测
2.4 模板存储温度对PCR扩增的影响
为了明确制备样本的保存时间和温度对于PCR扩增的影响,将DNA样本分别存放于-20℃和室温(25℃),14 d后再次进行PCR扩增。与新制备样本(图2)相比,-20℃存储样本对PCR扩增无明显影响(图3A),室温存放结果显示N2~N7和TE扩增效果不变,而N8、T50以及DDW出现了弥散(图3B)。

图3 制备的DNA样本在不同存储温度下的PCR扩增结果
2.5 优选方法
依据制备样本的PCR扩增效果、存储状况以及样本溶液的状态,确立了内生真菌ITS检测中模板DNA的快速制备流程:1.5 mL离心管中放入两个3 mm研磨珠,随后加入0.500 mol/L的NaOH提取缓冲液100 μL,在超净工作台中用牙签挑取少量菌丝于上述离心管中,采用组织研磨仪于25 Hz破碎2 min,瞬时离心后90℃金属浴中3~5 min,再次瞬时离心后加入300 μL的0.05 mol/L的Tris·HCl(pH 8.0),即为制备的PCR模板,-20℃或室温保存备用。
2.6 优选方法对68个属真菌的分类鉴定。
应用优选方法对本实验室保存的白僵菌以及分离于玉米、大豆、棉花、地瓜植株的772个菌株进行扩增测试,结果表明711个可产生有效扩增,62个无带或扩增较弱,扩增成功率为91.98%。Sanger测序结果显示706个为单峰,5个菌株测序图为双峰,序列经本地Blastn(NT)比对鉴定后分布于68属199种,图4展示了68个属的扩增结果,图5为其中一个样本的Sanger测序峰图。
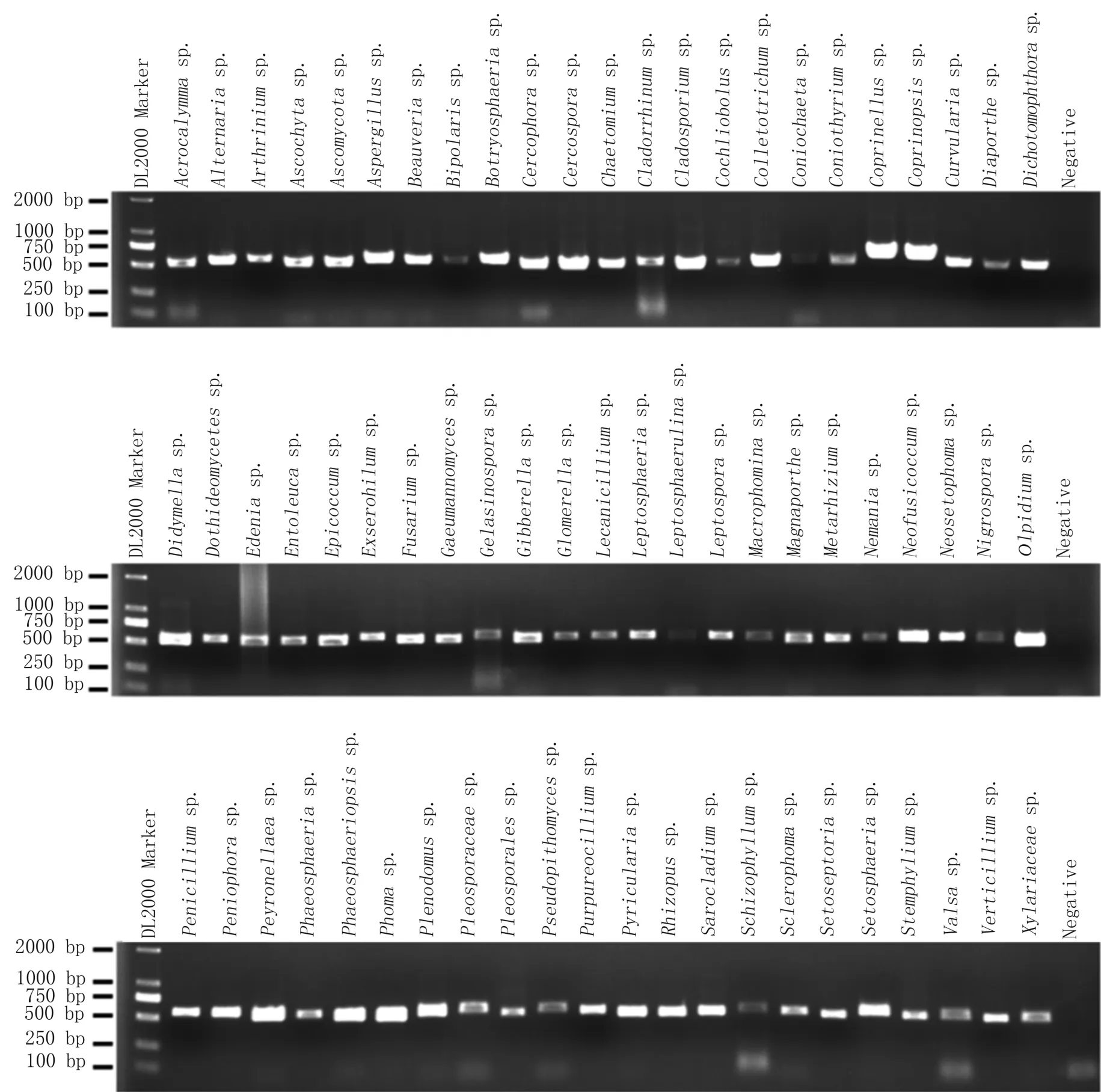
图4 优选方法对68个属真菌的PCR鉴定

图5 PCR产物的Sanger测序
2.7 核糖体大亚基引物扩增测试
为了明确ITS引物扩增失败的62个样本是否与模板制备有关,利用nLSU引物(LROR/LR5扩增条带大小939 bp、LR3R/LR5扩增条带大小327 bp、LR3R/LR7扩增条带大小782 bp)再次进行PCR扩增。由图6可知,3对引物均可产生清晰扩增,该结果进一步表明ITS扩增失败并非样本制备问题。

图6 nLSU核糖体3对引物在ITS扩增失败的8个随机样本中的扩增
3 讨论与结论
碱裂解法已广泛应用于植物DNA快速提取,该方法的最大优势在于DNA样本制备高效、经济、环保[37],然而用该方法提取真菌DNA鲜见报道。本研究改进了碱裂解法并对其在真菌DNA快速提取中的应用及适用范围进行了详细阐述,优化了提取流程,拓展了该方法在植物内生真菌PCR检测中的范围。
针对白僵菌的测试中,DNA样本粗提液(不稀释)和稀释后PCR的结果均证明,DNA样本溶液中NaOH的浓度大于等于0.250 mol/L时,可完全抑制PCR反应,而不高于0.125 mol/L可正常进行PCR扩增,这一结果与徐宗昌等[38]的一致,其原因可能为高浓度的NaOH(≥0.250 mol/L)pH值≥13.4(https://www.osgeo.cn/app/s1678),该pH值超出了PCR缓冲液的缓冲容量,导致PCR无法进行。DNA溶液不稀释直接进行PCR,低浓度的NaOH、T50、TE、DDW虽然无法彻底裂解白僵菌,但仍可以进行PCR扩增,其原因可能为高温导致部分DNA的释放,满足了PCR扩增对模板的要求,这为某些种属真菌的裂解产物不稀释直接进行PCR扩增提供了条件支持。但是由于NaOH浓度较低,其适用范围可能具有一定的限制,需要进行单独测试。DNA样本的存储试验表明,低浓度的NaOH、T50及DDW制备样本在储存过程中出现了部分降解现象,而高浓度的NaOH(N2-N7)和TE溶液扩增效果未发现弥散,其原因可能为高浓度的NaOH及微量的EDTA均可抑制核酸酶的活性。
3对nLSU引物PCR成功扩增,表明62个样本的扩增失败并非DNA样本制备问题,而是由于ITS1/ITS4引物匹配率不高。其片段长度测试则显示了该方法可进行1 000 bp以下的快速PCR检测,完全可以满足ITS间隔约600 bp测序的要求。
本研究利用0.500 mol/L的NaOH和0.05 mol/L的Tris·HCl(pH8.0)优化了内生真菌DNA快速提取方法,该方法简便快捷、所需样本极少、不需要液氮及手工研磨、不使用有机溶剂且室温下可长期储存,并成功应用于68个属199个种的内生真菌鉴定,极大拓展了碱裂解法在内生真菌PCR分类鉴定中的适用种属范围,可满足大规模快速真菌鉴定的需要。
