基于第一性原理的氢化锆热散射律计算分析
2022-01-27祖铁军汤勇强曹良志吴宏春
祖铁军,汤勇强,曹良志,吴宏春
(西安交通大学 核科学与技术学院,陕西 西安 710049)
因具有高氢含量和高抗辐照损伤能力,氢化锆(ZrHx)作为一种高质量的慢化剂被广泛应用于TRIGA等反应堆中[1]。氢化锆的热散射截面,特别是氢化锆中氢的热散射截面对反应堆中子学计算有着重要影响,获得高精度的氢化锆热散射截面是十分必要的。热散射律数据是计算热散射截面的基本数据。热中子散射受材料晶体结构的影响较大,不同氢含量的氢化锆会有不同的晶体结构,因此需根据氢化锆实际晶体结构计算其热散射律数据[2]。
在基于声子展开方法的热散射律数据计算模型[3]中,基本的输入参数是反映散射材料晶体结构特性的声子态密度。对于ZrHx中氢的声子态密度,1968年,Slaggie等[4]建立了ZrH2的中心力晶格动力学模型(CF模型),采用面心立方晶体结构来近似ZrH2的微四方晶体结构,然后通过拟合热容数据得到声子态密度。美国评价核数据库ENDF/B采用了CF模型。2005年,Mattes等[5]建立了Debye-Gaussian(DG)模型,采用参数化的公式来描述ZrHx中氢的声子态密度,认为氢的声子态密度由符合Debye分布的声学部分和高斯分布的光学部分组成。欧洲评价核数据库JEFF采用了DG模型。Zheng等[6]基于DG模型,以TRIGA反应堆的实验结果为基础,建立了ZrHx声子态密度参数标定方法。Malik等[7]建立了ZrH1.58中氢的三高斯模型。
上述模型的共同特点是采用参数化公式表示ZrHx中氢的声子态密度。基于热散射截面、反应堆的反应性等实验结果拟合得到声子态密度公式中的参数,不能反映ZrHx的真实晶体结构,会在热散射截面计算中引入误差。近年来,核材料研究领域通过第一性原理计算成功得到与实验吻合的ZrHx的晶体结构[8-9],计算中可得到相应晶体结构的声子态密度。与参数化声子态密度相比,第一性原理计算得到的声子态密度能较好地反映ZrHx的真实晶体结构。因此,本文利用第一性原理计算方法计算ZrHx中氢的声子态密度,然后通过核数据处理程序NECP-Atlas[10-11]计算其热散射律数据和热散射截面,分析不同方法获得的声子态密度对ZrHx热散射截面的影响以及对含ZrHx的TRIGA反应堆反应性的影响。
1 热中子散射截面计算方法
热中子散射的双微分截面σ(E→E′,μ)可表示为:
σ(E→E′,μ)=
(1)
式中:E为入射中子能量;E′为出射中子能量;μ为实验室坐标系下的散射角余弦;kB为玻尔兹曼常数;T为材料的温度;σcoh为束缚核的相干散射截面;σinc为束缚核的非相干散射截面;α为中子的无量纲动量转移量;β为中子的无量纲能量转移量;S(α,β)为热散射函数;Ss(α,β)为自散射函数。α和β分别表征中子散射前后角度和能量的变化量,可分别表示为:
(2)
(3)
式中,A为原子与中子的质量比。
对于固态晶体材料,原子在平衡位置附近做微小的振动。在这种情况下,晶体中原子间的相互作用力可认为正比于原子的位移,原子间的力可假定为简谐函数,称之为简谐近似。因此,热中子散射截面可用声子展开模型表示为:
(4)

对于ZrHx中的氢,在热能区有3种类型的散射:非弹性散射、非相干弹性散射和相干弹性散射。目前的评价核数据库中忽略ZrHx中氢的相干弹性散射项。因此,为保持与评价核数据库一致,以下只讨论非弹性散射和非相干弹性散射。
1.1 非弹性散射
对于非弹性散射过程,散射系统中至少有1个声子产生或者湮灭,因此,保留式(4)中n≥1项,且采用非相干近似[3],非弹性散射的双微分截面σine(E→E′,μ)可表示为:
(5)

(6)
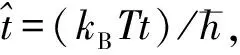
(7)
1.2 非相干弹性散射
对于非相干弹性散射,散射系统中没有声子产生或湮灭,只保留n=0项,所以非相干弹性散射截面σel,inc(E,μ)计算表达式为:
(8)
式中,2wE为德拜-沃勒积分,w由以下公式计算:
(9)
2 第一性原理计算及结果
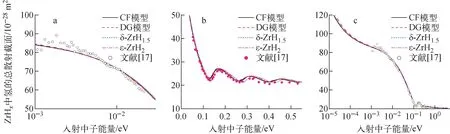
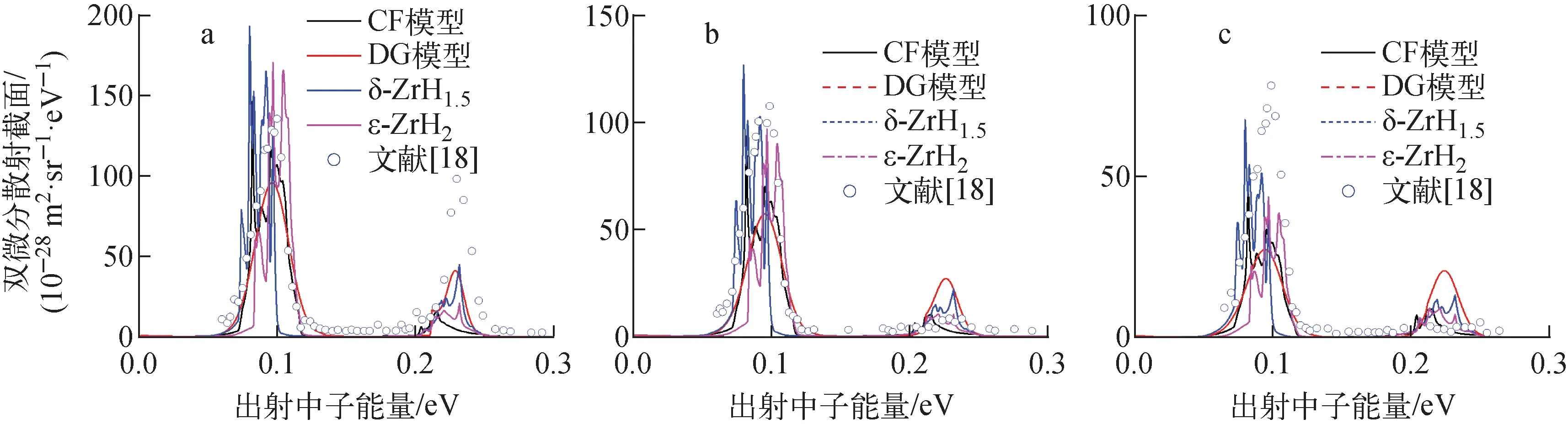
慢化剂材料ZrHx的氢含量一般在1.4 分别创建δ-ZrH1.510个原子(Zr4H6)的晶胞和ε-ZrH212个原子(Zr4H8)的晶胞,如图1所示;采用第一性原理材料模拟软件包VASP[12]对以上两个晶胞进行几何优化和能量评价,获得晶胞最低能量时的晶格常数;本文计算的δ-ZrH1.5的晶格常数为a=b=c=0.476 800 nm;ε-ZrH2的晶格常数为a=b=0.500 895 nm,c=0.445 270 nm。然后,采用晶格动力学计算程序PHONOPY[13]分别将δ-ZrH1.5和ε-ZrH2的晶胞扩展为2×2×2的超胞,然后用VASP中的密度泛函微扰理论(DFPT)方法计算超胞的原子间力常数。在VASP的计算中,用投影缀加波(PAW)方法描述电子离子相互作用,采用Perdew-Burke-Ernzerhof(PBE)公式中的广义梯度近似(GGA)来描述交换和关联势。布里渊区k点采用以Γ为中心的Monkhost-Pack方案取样,k点间距最小值为2π×0.04 Å-1。采用高斯弥散(Gaussian smearing)方法,并且弥散宽度取为0.05 eV。平面波基波的截止能量为500 eV。最后,根据VASP计算得到的原子间力常数,使用PHONOPY程序来计算声子态密度。 图1 δ-ZrH1.5(a)和ε-ZrH2 (b)的晶胞Fig.1 Unit cell of δ-ZrH1.5 (a) and ε-ZrH2 (b) 图2给出基于第一性原理计算获得的ε-ZrH2声子态密度和ENDF/B-Ⅷ.0中ZrH2的CF模型、JEFF-3.3中ZrHx的DG模型的声子态密度光学部分,并与Evans等[14]获得的ZrH2中氢的声子态密度光学部分的实验结果进行了比较。可见,采用第一性原理计算的ε-ZrH2声子态密度与实验结果吻合较好,很好地复现了在0.136和0.144 eV处的两个光学峰;在0.15~0.16 eV范围内,ε-ZrH2声子态密度与实验数据也有较好的一致性,两者的声子态密度都有一个明显的平台。CF模型在以上两个能量也出现了峰,但峰的位置与实验结果有偏差。DG模型则只有一个光滑的峰。 图2 ZrH2中氢声子态密度光学部分Fig.2 Optical phonon density of state of H in ZrH2 图3比较了第一性原理计算获得的δ-ZrH1.5和ε-ZrH2中氢的声子态密度、JEFF-3.3中DG模型、ENDF/B-Ⅷ.0中CF模型的声子态密度。ZrHx中氢的声子态密度分成两个区域:低能区的声学部分和高能区的光学部分。由于声学部分数值较小,图3中将声学部分的数值乘以100。DG模型假设声子态密度由Debye温度为20 meV的声学部分和高斯分布(在0.137 eV处达到峰值)的光学部分组成,因此声子态密度曲线是平滑变化的。δ-ZrH1.5声学部分的结果在形状和量级上与CF模型基本吻合,但峰值处的能量低于CF模型,两种模型出现峰值的能量点分别为0.015 eV和0.017 eV。对于ε-ZrH2的声学部分,主峰出现在20 meV处,与DG模型一致。在光学部分,δ-ZrH1.5的声子态密度振荡大于其他模型。Malik等[7]将ZrHx中氢的声子态密度光学部分被表示为1个三高斯模型,3个高斯分布的中心点分别在0.132、0.137和0.151 eV处,而δ-ZrH1.5的计算结果显示在0.135、0.139和0.152 eV处有3个峰值,这与三高斯模型一致。 图3 4种模型声子态密度的比较Fig.3 Comparison of phonon density of state among four models 基于以上获得声子态密度,利用核数据处理程序NECP-Atlas中sab_calc模块[15]计算了ZrHx中氢的热散射律数据,然后采用therm_calc模块[11]计算了热散射截面。将截面计算结果和国际原子能机构核反应实验数据库EXFOR[16]中的实验数据进行了详细对比,由于篇幅原因,下文仅给出部分结果。 图4示出能量范围为1.07×10-3~2.17×10-2eV、5.3×10-2~5.3×10-1eV和热能区的ZrH1.5中氢的总散射截面的计算结果及实验结果[16-17]。由图4a可看出,在此能量范围内基于不同模型的声子态密度计算的总散射截面无明显差异。由图4b可看出,ε-ZrH2、CF模型和DG模型的结果基本吻合,δ-ZrH1.5与其他模型计算结果存在差异。 图5示出入射能量Ei=0.232 eV、散射角θ=60°、90°和120°时ZrH2的双微分散射截面的计算值及实验结果[18]。由于双微分截面描述的是在某一入射能量下,中子在特定能量、角度出射的概率,对晶体结构较为敏感,所以相对于图4a、b展示的总散射截面,不同的声子态密度计算模型体现出明显的差异,且由于δ-ZrH1.5的结构实验样品不一致,基于δ-ZrH1.5声子态密度计算的双微分散射截面有明显偏差。基于ε-ZrH2的声子态密度计算的双微分散射截面与实验值吻合最好,其次是CF模型和DG模型。 能量范围:a——1.07×10-3~2.17×10-2 eV;b——5.3×10-2~5.3×10-1 eV;c——热能区图4 ZrH1.5中氢在不同能量范围的总散射截面Fig.4 Total scattering cross section of H in ZrH1.5 in different energy regions a——Ei=0.232 eV,θ=60°;b——Ei=0.232 eV,θ=90°;c——Ei=0.232 eV,θ=120°图5 ZrH2的双微分散射截面Fig.5 Double differential scattering cross section of ZrH2 利用基于不同声子态密度获得的热散射截面,对国际临界安全基准题库ICSBEP[19]中含ZrHx的TRIGA反应堆基准题进行了计算,计算的问题列于表1。计算时ZrHx中锆采用的数据为:ENDF/B-Ⅷ.0采用其中给出的ZrHx中锆的热散射律数据;JEFF-3.3中无锆的热散射律数据则采用自由气体模型;本文采用不同晶体结构分别获得ZrHx中锆的热散射律数据用于相应计算。研究中发现ZrHx中Zr的热散射律数据对结果影响很小。其他所有核素的截面数据均来自ENDF/B-Ⅷ.0。采用蒙特卡罗程序MCNP进行中子学计算,计算中有效增殖因数keff的统计偏差控制在±10 pcm以内。计算的keff与实验值比较列于表2。表2中:ICT003和ICT013基准题中ZrHx处于δ相,所以计算中使用了基于δ-ZrH1.5声子态密度的截面;HCM003基准题中ZrHx处于ε相,采用基于ε-ZrH2声子态密度的截面进行计算。 表1 ICSBEP临界基准题中氢/锆原子比Table 1 H/Zr atom ratio in ICSBEP criticality benchmark 由表2可见,CF和DG模型对所有基准题结果比较吻合。基于第一性原理声子态密度的热散射截面对计算结果有明显改善,特别是对于含有δ-ZrH1.5的ICT003和ICT013基准题。与CF模型相比,采用第一性原理计算的声子态密度可使ICT003_02基准题偏差绝对值最大减少332 pcm;与DG模型相比,也可使ICT003_02基准题偏差绝对值最大减少448 pcm。 表2 计算的keff与实验值比较Table 2 Comparison between calculated keff and experiment value 对于HCM003系列基准题,本文计算结果与实验值的偏差控制在100 pcm以内。 为进一步展示不同模型对中子能谱的影响,对ICT003_02基准题中A1燃料棒的中子能谱进行了对比,如图6所示。由图6可见,CF模型和DG模型的中子能谱无明显差异,而本文结果相对于前两者,在0.08~0.16 eV范围内中子通量有所增加,出现了能谱硬化。 图6 ICT003_02基准题中A1燃料棒的中子通量Fig.6 Neutron flux of A1 fuel rod in ICT003_02 benchmark 采用第一性原理计算了氢化锆的δ-ZrH1.5和ε-ZrH2两种晶体结构中氢的声子态密度,以此计算了ZrHx中氢的热散射律数据和热散射截面,然后将热散射截面应用到国际临界安全基准题中含ZrHx的TRIGA反应堆的中子学计算,分析了不同声子态密度计算方法对热中子散射截面、TRIGA反应堆反应性的影响。结果表明:基于实际晶体结构,由第一性原理计算得到的声子态密度可给出更精确的热中子散射截面数据;与ENDF/B-Ⅷ.0和JEFF-3.3评价核数据库中模型相比,基于第一性原理获得的声子态密度可明显提高TRIGA反应堆反应性的计算精度。


3 热散射截面及TRIGA反应堆计算结果
3.1 热散射截面
3.2 TRIGA反应堆计算结果



4 结论
