冗余分析在微生物生态学研究中的应用
2022-01-26任玉连董醇波邵秋雨张芝元梁宗琦韩燕峰
任玉连,董醇波,邵秋雨,张芝元,梁宗琦,韩燕峰
(贵州大学 生命科学学院生态系真菌资源研究所,贵州 贵阳 550025)
冗余分析(Redundancy analysis,RDA)是多元回归模型的延伸,其用排序的方法阐述群落生境或其中某一个生态因子随样地生境的变化。由于其能快速获得解释变量与响应变量间的关系,近年来在生态学领域的研究中受到越来越多的重视。由生物或非生物介导的地上或地下等复杂的生态过程中,影响因子绝不是单一的,其作用也不可能独立。冗余分析经过对特征值进行一系列分解筛选,可实现有效简化目标变量个数的目的,进而将物种与环境因子的关系直观地体现在同一坐标轴上,最终获得解释变量与响应变量之间的关系,获得一个或多个主导因素。此外,冗余分析还能独立保持各个解释变量对响应变量的贡献率,同时对具体指标解释能力大小及排序可靠性进行定量描述,以及具有识别环境变量组合的主要选择性梯度的优势。因此,使用冗余分析能快速有效地解决应用研究中的多因子影响贡献率排序问题。
目前,许多学者使用CANOCO软件进行冗余分析。该软件在解释大尺度宏观生物群落结构、动植物区系与环境之间的关系,检验假设冲击对环境和其生物群落所造成影响的程度,以及在分析不同的生态系统和生态毒理学等领域具有广泛的运用。自1985年CANOCO 1.0发布以来,已发布了6个版本,现广泛使用的是CANOCO 5。该软件包含了经典RDA冗余分析、梯度分析、分类、多元回归、相似性度量和实验设计等排序方法。由于其操作简单、功能齐全,是生态学及相关领域多元数据排序分析的流行软件之一。
冗余分析的应用已从宏观生态分析,如与水生动植物、鸟类、植被、土地利用类型及景观格局分布的关键环境因子筛选,逐渐被运用到微生物生态学及其与人类相关疾病等领域。在微观土壤微生物生态学研究中,冗余分析被用来进行微生物群落结构、组成、多样性及功能等方面主控因素的提取。在这类冗余分析时,一方面需要考虑单个因素对微生物的影响;同时,也需要分析多个因素对微生物的影响程度。
冗余分析中的数据选择是灵活分析不同解释变量与响应变量关系的核心。在微生物生态学研究中,多数学者将微生物群落结构及多样性作为一组变量,而把环境因子作为另一组变量进行冗余分析,这样既能很好地揭示环境因素对微生物群落结构及多样性整体的影响,也能很好地揭示环境因子对各物种的影响贡献大小。如Yang等为弄清土壤微生物群落结构的关键作用和潜在机制,基于分类距离和系统距离对细菌群落进行冗余分析。Cao等基于Biolog生态平板数据,以确定土壤生化特性与细菌群落组成和功能多样性间的关系,将土壤环境变量作为解释变量,而将不同处理下微生物代谢功能频率分布变化,作为响应变量进行冗余分析。此外,Wang等则将针叶林和阔叶林的凋落物特征、土壤性质作为解释变量,土壤微生物群落则为响应变量,进行冗余分析后,快速获得了土壤特性、凋落物性质对微生物群落结构总变异的解释量排序。因此,在进行冗余分析时,需针对不同的研究目的,谨慎选取解释变量与响应变量是正确获得冗余分析结果的关键。
1 冗余分析概念
冗余分析(Redundancy analysis,RDA)是一种回归分析结合主成分分析的排序方法,能对多个解释变量(1…)的多个响应变量(1…)进行回归分析,将解释和响应两组变量的数据集通过双标图显示二者间的相关性变异程度。如在一元线性回归分析中,每一个物种的多度通过对应的解释变量进行回归分析:
=++
(1)
是样方物种的多度,是样方的已知解释变量。将物种的多度和解释变量定义为一种线性关系,和分别是这一直线的截距和斜率。通过环境变量的加权总和约束。以2个环境变量为例:
=1+2
(2)
可获得最优权重和,其权重值为典型相关系数。将(2)代入(1)中可得:
=+1+2+
(3)
通过物种数据和环境数据1获得模型中的物种参数和(1,…,m)以及权重值和,即
1=,2=
(4)
将模型(3)改为:
=+11+22+
(5)
综上,冗余分析是一个对所有物种同时进行多元回归的模型,既是一种约束性的主成分分析,也是一种约束性的多元多重回归。
此外,冗余分析可将一维模型推广到多维。以两维模型为例:
=+11+22+
(6)
为第个物种的得分,是第个样方在第(=1,2)排序轴上的得分。样方得分通过(7)进行约束:
=11+22
(7)
为第个环境变量在排序轴上的典型相关系数。将(7)代入(6)可得回归系数受约束后的多元回归模型。以代表回归系数为例:
=11+22
(8)
该模型是回归双标图的基础,通过(6)可看出样方得分和物种得分共同形成物种拟合值的双标图。
2 基于CANOCO 4.5进行冗余分析操作
CANOCO 4.5软件主要包括Canoco for Windows、WcanoIMP、CanoDraw、CanoMerge和PrCoord五大模块。其中,Canoco for Windows用来指定需分析的数据和排列模型;WcanoIMP将Excel等形式的数据转化为CANOCO能识别的*.dta形式;CanoDraw进行排序图的生成,以及生成不同类型的等值线和回归模型;CanoMerge是合并CANOCO能识别的*.dta类型数据文件,可将数据文件以带制表分隔符的文本形式输出,具有滤掉低频率物种的功能;PrCoord对特定数据集进行主坐标分析以及冗余分析。
2.1 软件安装
下载国际通用软件包CANOCO 4.5,解压并按照提示安装程序updt_456.rar安装软件。
2.2 原始数据
输入数据时应遵循样方名和变量名命名规则,一般来说字符不得超过8个;建议使用简单的数字、字母、点、连接符和空格结合进行命名;在Excel中将物种变量和环境变量按照样方名称和变量名称命名格式输入需要分析的数据。确保每一行为一个样本,每一列为一个物种(或变量)。如果物种变量和环境变量之间有数量级差别,则需要对数据进行数据标准化处理(如对数转换、Z-Scoring等)。
2.3 数据转换
CANOCO 4.5版本很好地解决了数据文件过于复杂和格式问题。即用WCanolomp将两组数据转换为CANOCO能识别的格式文件。打开已备好的原始数据,分物种变量和环境变量拷贝后,访问WCanoImp.exe程序。如图1对话框所示,确定所选选项正确后,单击保存按钮继续,指定要生成的文件夹和存储位置,用于正在生成的数据集。同时,WCanoImp程序运行的另一个对话框会显示成功创建。此时,Excel数据已转化为CANOCO可识别数据(后缀以*.dta命名,如“spe.dta”“env.dta”)。
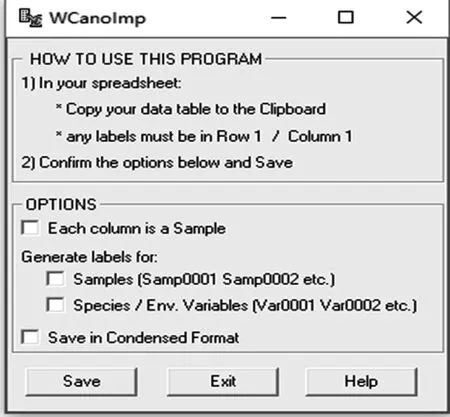
图1 WCanoImp程序窗口Fig.1 The program window of WCanoImp
2.4 排序方法的选择
CANOCO 4.5版本能快速进行去趋势分析(Detrended correspondence analysis,DCA),它是判别使用RDA或典范对应分析(Canonical correspondence analysis,CCA,也是一种分析生物群落与环境因子间相互关系的一种方法)的判断依据。利用Canoco for windows>New project>Available Data>Next>Data files>Browse指定要分析数据文件>下一步>DCA>Finish Options>点击“Project View”窗口的“Analysis”>分析结果即可在“Log View”窗口中查看。在得到4个排序轴的梯度长度后,根据DCA分析结果中的Lengths of gradient数值大小判断选取RDA或CCA方法。梯度长度<3时,使用RDA线性响应模型(图2);梯度长度介于3~4之间,RDA与CCA分析均可;梯度长度>4时,选择CCA单峰响应模型进行分析。

图2 Log View 窗口中分析结果Fig.2 Analysis results in Log View window
2.5 RDA模型建立
在CANOCO 4.5菜单栏上依次点选:File>New project>Available Data界面,选择“Species and environment data available”> Next,将上述“spe.dta和env.dta”文件对应添加到“Data”Files”界面的前两列中,最后一列是输出“*.sol”文件>下一步> RDA>Scaling Linear Methods>Symmetric>Divide by standard deviation>Finish Options>Canoco Project,点击“Project View”窗口中“Analysis”> 保存生成的“*.Log”文件,可在“Log View”窗口中查看分析结果。在RDA分析中,需要对环境变量选择手动筛选(Manual selection),在Project>Analysis中弹出的对话框中,依次检验环境变量的解释度(Test variable),根据P值大小,选择需要的变量。
2.6 RDA模型作图
从上一步骤中的Project view >激活CanoDraw.exe>Create>Biplots and Joint Plots>Species and env.Variables / Species and samples / Samples and env.variables即可对物种以及环境变量/物种以及样品/样品以及环境变量作图。最后将生成的RDA图经线条、颜色、字体等调整后保存并输出。如图3所示,排序轴以土壤真菌被理化因子的线性组合解释量的多少先后出现(排序轴1,2,3和4……n),前四轴通常会占到解释量的较大部分,因此在排序图中一般只选取前四轴中的两个进行制图。
2.7 冗余分析排序图的解释
图3为真菌属分类水平与土壤环境变量间的冗余分析结果。其中,黑色实心箭头代表各类真菌属;黑色空心箭头代表理化因子;黑色实心箭头连线与黑色空心箭头连线之间的夹角代表某类真菌属与某理化因子之间的相关性,用夹角的余弦值表示;即箭头连线与排序轴的夹角表示真菌属与理化因子间的相关性。夹角接近90°为接近正交,表明真菌属与理化因子之间的相关性很小,二者间几乎不存在影响(图3b:OM与镰刀菌属)。夹角小于90°为锐角,表明真菌属与理化因子之间存在正相关(图3b:AP与曲霉属);锐角角度越小,则正相关性越大(图3a:AP与腐霉属)。夹角大于90°为钝角,表明真菌属与理化因子之间存在负相关(图3b:pH与粪壳属);钝角角度越大,则负相关性越大(图3a:TN与青霉属)。黑色空心箭头所处的象限代表理化因子和排序轴的正负相关性;黑色空心箭头连线在排序轴上投影的长短表示某个理化因子与排序轴之间相关性大小,投影长度越长,则相关性越大。图3冗余分析显示,土壤不同理化因子影响其不同真菌类群的生存繁殖。0~20 cm与20~40 cm土壤中,第Ⅰ轴和第Ⅱ轴真菌群落特征—土壤理化因子关系累计解释量分别高达65%和91%以上(图3 a、b)。0~20 cm土层,有机质、全氮和速效磷等理化因子对50%以上的菌群具有促进或抑制作用,是影响真菌群落结构的重要因子,而20~40 cm土层影响其真菌群落结构的是速效钾、全氮、容重、自然含水率和pH等理化因子。说明土壤理化因子在决定土壤真菌群落结构分布中起着重要作用。

注:a.0~20 cm土层土壤真菌属与环境变量关系;b.20~40 cm土层土壤真菌属与环境变量关系。图3 基于CANOCO 软件RDA分析的物种与环境变量关系图[13]Fig.3 Relationship between species and environmental variables based on RDA of CANOCO software
3 冗余分析在微生物生态学研究中的应用
冗余分析作为生物与环境变量关系的一种测度和类型,与回归分析、相关分析、主成分分析、结构方程模型以及可视化网络等分析方法目的相同,均是寻找解释变量与响应变量之间的关系。由于冗余分析的简洁、快速和可视化的特点,在微生物生态学研究领域中,已越来越多的受到研究者的关注。
3.1 微生物群落组成及多样性与环境因子间关系分析

3.2 碳源代谢分析
微生物代谢多样性能够反映微生物群落的生态特征。由于土壤微生物的碳源代谢能力受土壤性质、植物多样性、凋落物组成等因素影响,但是对于不同生境中的研究,不同学者选取的解释变量和响应变量具有一定差异。如王芸等基于含水量、沙砾和容重为解释变量,以微生物碳源代谢为响应变量的分析结果显示,含水量、沙砾和容重显著影响土壤微生物碳源代谢功能,分别能解释10.5%、8.8%和7.2 %的变异。土壤pH值被用来解释微生物代谢能力的重要变量之一,因为随着土壤pH的减小,土壤代谢强度显著增加。此外,电导率、全氮和碱解氮能显著促进微生物的碳源代谢活性,土壤盐度则显著抑制了土壤微生物的碳源代谢活性,也被选作冗余分析时的解释变量,特别在不同经营模式林分土壤微生物的碳源利用研究中被广泛使用。此外,由于土壤理化性质、植被多样性和水热条件等能影响土壤微生物代谢作用释放分泌某些酶类,因此土壤酶活性被用来作为冗余分析中重要的响应变量之一。如任玉连等使用样地微气候(年均气温、年均降水量)、植被多样性指数(Margalef丰富度指数、Shannon)样性指数、Pielou均匀度指数及Simpson指数)和土壤理化性质(土壤密度、土壤温度、含水量、pH、有机碳、全氮、水解性氮、全钾、速效钾、全磷、有效磷及C/N)与土壤酶活性(脲酶、蔗糖酶、酸性磷酸酶、多酚氧化酶、过氧化氢酶)进行冗余分析,最终获得单一环境因子对土壤酶活性影响的重要性排序。曹聪等认为使用冗余分析排序图可以看出理化性质(温度、水分、碳、氮和磷)对酶活性有不同程度的影响。因此,使用冗余分析可以快速获得环境因子与微生物碳源代谢能力间的关系,但在选取解释变量的过程中,应考虑生境条件及试验条件的差异,尽可能获得更为丰富解释变量数据进行冗余分析。
3.3 核心微生物组及功能分析
核心微生物组研究最早开始于人体的体表面和内部共栖、共生微生物组的部分成员,现已广泛涉及动物、植物、土壤、水体以及废水处理系统微生物组中有关的成员,它们是微生物组的基本组成及功能的关键部分。由于核心微生物组在分类学上呈现的生物多样性特性,与相关物种共存机制以及在群落功能中的作用相关,因此多数学者仅关注核心微生物组间的关系。目前主要基于分子技术用MetaCoMET、COREMIC、PhyloCore、BURRITO等分析工具综合界定核心微生物组,但核心微生物组与环境变量间的关系较少被关注。Mukhtar等在门水平上以古菌、细菌和真菌为响应变量;以头孢氨苄、温度和水力保留时间为解释变量,分析揭示环境因素对细菌、真菌和古细菌群落结构的影响。此外,冗余分析也被用于寻找功能微生物(降解有机污染物的微生物菌群)中的中枢(hub)菌属。在揭示绿肥处理影响微生物相关功能基因的重要因素的冗余分析中,则以微生物功能基因相对丰度为响应变量,以碳、氮、磷、铁、硫、pH以及植物激素为解释变量。此外,也有学者采用冗余分析对盐胁迫下的核心微生物分类菌群进行研究。
3.4 微生物谱系地理
生态系统中碳、氮、磷等循环可能将生物地球化学模式与生理限制联系起来,是微生物多样性和营养限制的主要驱动力和关键预测因子。由于不同物种的分布模式差异巨大,探索哪些因素是影响物种分布模式、遗传多样性和种群结构及群落扩散的关键因素尤为重要。因此,冗余分析也与其他分析工具相结合,从而实现不同的研究目的。如冗余分析与结构方程模型结合,用来揭示不同区域水体养分特征(铁、锰、化学需氧量、营养盐和酸碱度)对藻类群落影响的主要环境因子。运用冗余分析验证纬度生物地理模式,并描述生物地理模式形成因素和关键分单元类群(Keystone taxa)。通过冗余分析解释低和高核酸含量细菌的丰度和细胞计数特征随地理距离和环境变量(pH、温度、盐度、经度、纬度、海拔、溶解氧、电导率、叶绿素-a)的变化,结果表明细胞计数特征和丰度与环境因素显著相关,且pH是驱动低和高核酸含量细菌变化的主导因素。这些结果显示了地理距离和环境改变是影响微生物地理分布的主要因素。
3.5 病原菌耐药性的环境因素分析
2020年新冠肺炎疫情危机,是一场人与微生物病毒的无声“争斗”,这种微生物给人类的健康和生存带来了巨大的威胁,生物安全问题已备受全球关注。微生物是导致人类传染病流行的最重要因素之一。近年来,人类在疾病的预防和治疗方面取得了长足的进展,但现代社会广谱抗生素的普遍使用已使许多菌株发生变异,导致耐药性的产生,新微生物感染不断发生。抗生素耐药基因在环境中的扩散已成为日益严重的健康风险。因此,基于冗余分析的便捷快速等优点,冗余分析可能在寻找影响微生物耐药性环境因素方面具有广阔的应用前景。如基于抗生素、金属、镁元素和细菌为环境变量,以精氨酸为响应变量,冗余分析发现,抗生素、金属、镁元素和细菌分别解释了微生物群落中精氨酸变异的0.7%、5.7%、12.4%和21.9%的贡献率。此外,Guo等以OTC(土霉素)和Cd(镉)等为解释变量,以抗生素耐性基因和微生群落为响应变量进行冗余分析,获得OTC和Cd等因素对微生物抗生素耐药性基因变化的解释率。Zhang等基于冗余分析揭示添加镉对土霉素污染土壤中精氨酸转运、微生物群落和人体病原菌的影响。由于影响人体微生物菌群的因素众多,因此,在开展这一领域的研究时,获得的解释变量越多,采用冗余分析能快速获得不同解释变量的贡献率,可能其实际运用价值更高。
4 展望
冗余分析结合环境矩阵和物种矩阵可直观、明确、有效地揭示环境因子与物种间相关程度的大小,是一种经典、成熟的有效方法,在传统生态学各领域研究中具有较为突出的优势。由于这种分析方法操作简单、快捷,在处理微生物组产生的巨量数据面前,能较快地找到环境与物种间的复杂关系并对变量之间的关系作出直观的生态解释,凸显了它的优点和优势。故已在不同生态系统中微生物组的组成、结构、功能、生物多样性及碳源代谢方面皆展现了广阔的应用前景。今后,冗余分析解析方法主要从以下几个方向发展。
4.1 解释变量的高度选择
微生物生态研究中使用冗余分析时,解释变量的指标选取,成为影响研究结论的核心因素。由于环境因素复杂多样、群体大、且各群落间存在许多促进和抑制的关联,为能在众多解释变量中精准获得主控因子。在主影响因素评价中,仅选择少量指标,对主要影响因子很难作出准确合理的抉择。因此,在不同的研究中,应围绕核心的研究目标,谨慎选取解释变量,并尽可能多地获得解释变量个数,以获得更为真实的研究结果。此外,在数据有限的情况下,如何快速高效地找出具代表性的环境变量因子也应是未来研究的重要内容之一。
4.2 进一步改进冗余分析方法
随着分析软件的不断发展,新的排序方法不断出现,为能满足相关生态学研究人员对于多元数据深入挖掘分析的需求。还需对该软件系统作进一步改进。为减少研究人员因软件模块多而增加分析工作量负担,需整合该软件使其不再有分离的模块,改进为集数据管理、分析和绘图为一体的单一程序。数据输入成功与否对进行下一步往往是初学者最大的障碍,需简化数据输入,直接导入数据即可进行分析是未来发展的趋势所向。而在最新版本中已实现并提供一套完善简单的绘图工具,简化方差分解和显著性检验的操作步骤,增加单个解释变量的显著性检验功能,增加群落内类群功能性状与谱系关联分析等。因此,对于大部分病原菌、病毒未知因素的整个生态环境而言,该分析软件具有实际应用价值,仍需进一步改进发展。
4.3 寻找病原菌、病毒主控因子
微生物的研究对于人类理解整个生物圈的能量流通和物质循环、了解微生物在整个生态系统中的功能、挖掘有用的潜在微生物资源以及保护生物安全方面有着非常重要的意义。目前,物种的功能性状与谱系分析是生态学研究的热点之一。冗余分析法分析功能多样性从探究陆地、水生生态系统中包括植物、动物、菌类,并已过渡至生物安全有关的疾病研究等不同营养级的生物体在群落和生态系统中的功能及其范围。但在微生物相关疾病研究中应用较少,病原菌、病毒与环境因素间的影响关系尚不清楚。尤其是研究暴露于环境中的病原微生物的功能性状、核心群组、谱系地理分布与各因素的关系,有望为解决该分析对寻找微生物领域潜在致病菌的主控影响因子提供极具价值的线索,为流行疾病的预防提供参考。如,哪些影响因子可以抑制或促进病原菌或病毒的扩散和变异,这些结果可能对临床试验提供一定的参考意义。根据基因、遗传、性状与环境数据探索这些物种的致病因素将成为未来的发展趋势。
4.4 多分析方法的有机结合
要实现全面,准确的微生物与环境间影响关系的评价,任何一种单一的分析软件都无法完成。尽管冗余分析方法提供了快速获得解释变量与响应变量间关系的解决方案,但该方法仍属于单一分析方法,其核心算法模型较为单一,可能获得的理论结果与实际情况具有一定的差异。因此,随着生态学处理数据相关软件功能的升级和完善,需综合考虑使用多软件(R语言、Python)、多分析方法(网络分析、结构方程模型)与冗余分析的结果进行对比分析,进而获得更为真实的结果。
