乌鲁木齐市活禽市场环境源H5N8亚型高致病性禽流感病毒的遗传进化分析
2022-01-25贠丰泽阿木提喀日马木提常娜娜苏斌杰毕玉海马正海
贠丰泽,阿木提喀日·马木提,张 成,2,常娜娜,苏斌杰,毕玉海,2,马正海*
(1.新疆大学 生命科学与技术学院/新疆生物资源基因工程重点实验室,新疆 乌鲁木齐 830046;2.中国科学院 微生物研究所/病原微生物与免疫学重点实验室/流感研究与预警中心,北京 100101)
对高致病性禽流感病毒(highly pathogenic avian influenza virus,HPAIV)等重大传染病病原的监测和风险评估是传染病防控的关键和生物安全的重要组成部分。近年来,新疆频繁暴发禽流感(avian influenza,AI)疫情,在新疆展开禽流感病毒(avian influenza virus,AIV)监测和病毒溯源等研究将为AI防控工作提供科学依据,具有重要的现实意义。2010年,在江苏活禽市场家鸭中检测到H5N8 HPAIV[1],随后于2014年1月在韩国一养禽场再次被发现,并在韩国快速传播[2],目前,H5N8亚型HPAIV已在全球广泛传播,在日本、俄罗斯、德国、匈牙利、意大利、荷兰和英国等欧洲国家以及美国均已检出该病毒[3-5]。大量的研究表明,活禽市场(live poultry markets,LPM)是多种AIV亚型共存的“大熔炉”,从LPM的禽类双腔拭子和外环境中几乎检测到了AIV所有亚型,LPM在AIV传播、进化、突变、重配等方面也均发挥了重要作用[6-9]。同时,LPM也是人感染AIV的主要源头,据流行病学统计,感染HPAIV的患者大多数都有活禽暴露史[9-11]。为此,对LPM家禽和环境进行AIV监测是AI防控的重要环节。本研究定期采集乌鲁木齐市活禽市场外环境样品进行AIV分离鉴定,对分离获得的2株环境源H5N8 AIVs进行全基因组测序以及遗传进化和分子特征分析,以探讨乌鲁木齐市H5N8 HPAIV的来源及其潜在的致病性,为AI防控提供参考。
1 材料与方法
1.1 样品2016年10月至2018年12月于乌鲁木齐市某LPM定期采集环境混合样(笼具、笼具下粪便、屠宰案板、家禽饮用水和食物等),每月1次,每次采集2~3份。样品贮存于含多种抗生素的DMEM缓冲液中,于-20℃便携式冰箱运回实验室,-80℃保存备用。
1.2 主要试剂病毒RNA纯化试剂盒(BSC58S-2D)和PCR产物纯化试剂(BSC58S2C)购自杭州博日科技有限公司;一步法RT-PCR试剂盒(RR055A)和DNA Marker购自大连TaKaRa公司;DNA琼脂糖凝胶回收试剂盒购自天根生物科技有限公司;反转录试剂盒为日本TOYOBO产品(FSQ-201);高保真PCR试剂盒为美国MCLab产品。
1.3 AIV的分离样品管经30 s震荡、7 000 r/min离心5 min后,取100 μL上清接种于9~11日龄SPF鸡胚尿囊腔中。37℃培养鸡胚,24 h后每隔12 h 观察鸡胚死亡情况,弃去24 h内死亡的鸡胚,收取24 h后死亡的鸡胚或72 h未死亡的鸡胚尿囊液,经红细胞凝集试验检测为AIV阳性的尿囊液于-80℃保存备用。
1.4 AIV核酸提取从AIV阳性尿囊液中提取AIV核酸,具体操作按病毒RNA纯化试剂盒说明书进行。
1.5 AIV的鉴定以病毒RNA为模板反转录获得cDNA,继而以AIVPB1通用引物Flu-pan-PB1-F(ACIGGAGACAAIACNAAATGGAATGA)和Flu-Pan-PB1-R(ACTGTTGACAGCATITTNAACATNCCC)进行PCR扩增,扩增参数为98℃ 2 min;98℃ 10 s,55℃ 15 s,72℃ 10 s,35个循环;72℃ 7 min。最后用1.2%琼脂糖凝胶电泳检测PCR产物。
1.6 AIV全基因组扩增及测序对AIV阳性样品进行全基因组扩增和测序,以AIV通用引物MBTuni-12(ACGCGTGATCAGCAAAAGCAGG)和MBTuni-13(ACGCGAGATCAGTAGAAACAAG-G)进行RT-PCR,扩增AIV全基因组8个基因片段,反应参数为50℃ 30 min;94℃ 2 min;94℃ 30 s,55℃ 30 s,72℃ 150 s,35个循环;72℃ 7 min。1.0%琼脂糖凝胶电泳检测扩增产物,扩增产物回收后送华大基因进行二代测序。
1.7 AIV基因序列分析分离株基因序列在GISAID和GenBank数据库比对,获取与AIV 各基因片段相似性较高的病毒株序列为参考序列,以MegAlign 软件分析病毒各基因的同源性和突变,以MEGA-X的Maximum Likelihood法构建系统进化树,用Meg Align软件分析基因突变。
2 结果
2.1 病毒的分离、鉴定和基因组测序共采集LPM外环境样品75份,其中2016、2017和2018年分别为7,35,33份,从3份样品中分离到病毒,经测序以及HA和NA序列分析表明,其中2株为H5N8亚型,分别由2016年10月12日和12月18日的样品中分离;1株为H9N2亚型,自2017年3月样品中分离。本试验对2株H5N8 AIVs进行全基因组测序以及遗传进化和分子特征分析,2株环境源H5N8 AIVs间基因序列一致性分析表明,8个基因片段中MP、NA和NS的一致性较高,分别为99.2%,99.9%,100.0%,其他5个基因的一致性较低,为85.9%~98.1%,2株病毒存在差异,分别命名为A/Environment/Xinjiang/001/2016(H5N8)和A/Environment/Xinjiang/007/2016(H5N8),简称XJ/Env1/2016(H5N8)和XJ/Env7/2016(H5N8),其全长基因组8个片段的序列已提交至NCBI数据库,登录号为MW109845-109860。
2.2 AIV序列比对和遗传进化分析分离病毒各基因片段经BLAST比对,表1列出了与分离株各基因一致性最高的部分毒株。XJ/Env1/2016(H5N8)的8个基因片段与2016年底至2017年初自西藏、内蒙古和江苏等地区外环境中分离的H5N8 AIVs一致性最高,为95.21%~100.00%;另外NA和NS与2010-2012年华东地区家鸭中分离的H5N8 AIVs一致性亦为最高,分别为99.93% 和100.00%。XJ/Env7/2016(H5N8)的PB2与2011年山东野鸭中分离的H5N1 AIVs一致性最高,其余7个基因均与2016年底至2017年初自西藏和内蒙古外环境中分离的H5N8 AIVs一致性最高,PB1和PA分别为99.08%和99.95%,其余5个基因均为100.00%;另外HA、NP、NA、MP和NS5个基因与2010-2012年华东地区家鸭中分离的H5N8 AIVs一致性亦为最高,均为100.00%。
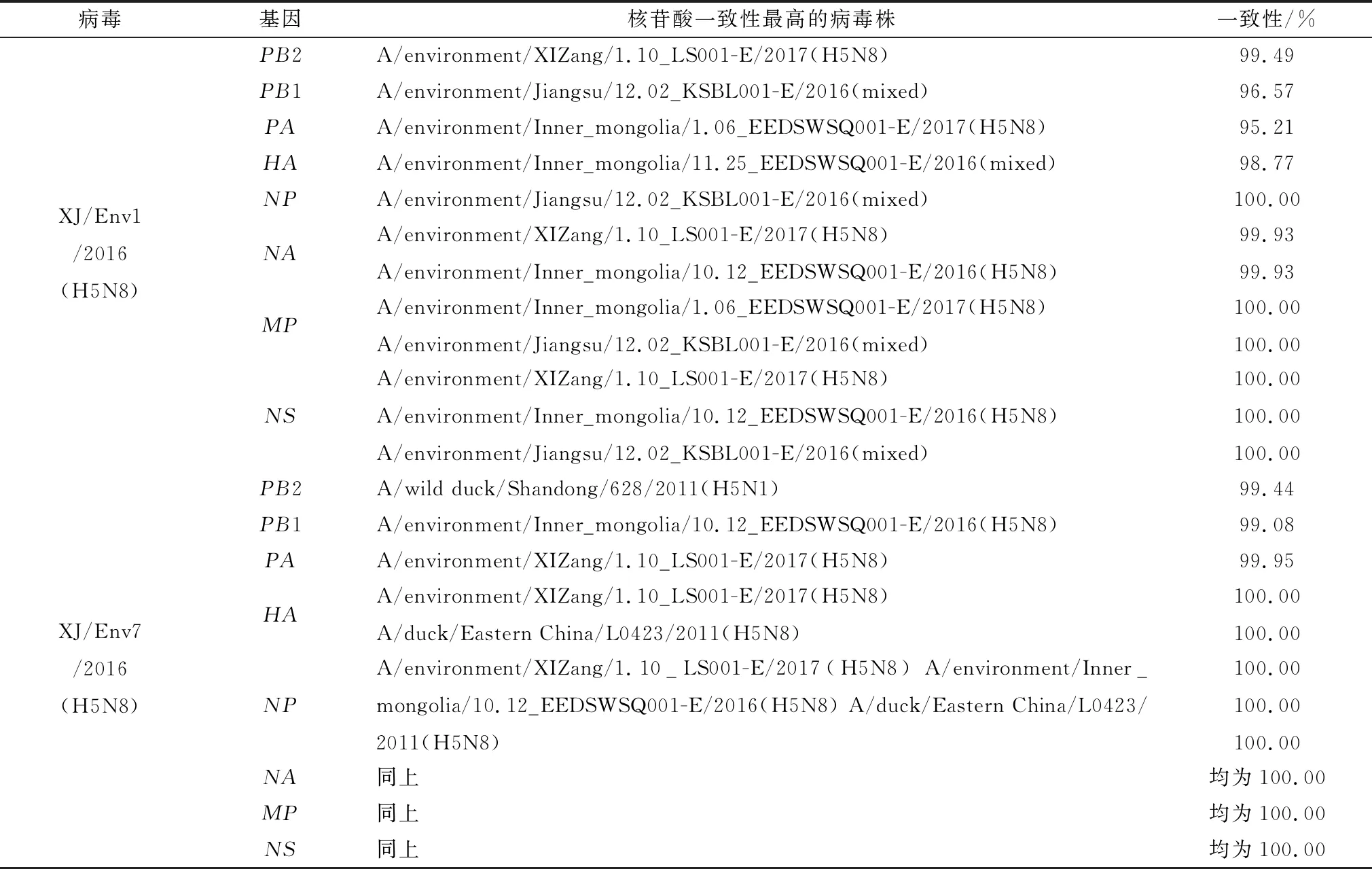
表1 与H5N8 AIVs分离株各基因核酸一致性最高的病毒株
H5N8 AIV分离株外部和内部基因系统进化分析如图1和2所示,根据HA的进化分析,2株病毒均属于Clade 2.3.4.4 Group A,2株病毒的NA和NS聚在一起,其余6个基因均聚在不同分支。同时,2株病毒各基因的进化关系存在明显共性,除XJ/Env7/2016(H5N8)的PB2与2010-2012年自江苏等华东地区家鸭中分离的H5N8 AIV聚在一个分支之外,XJ/Env7/2016(H5N8)其余的7个基因以及XJ/Env1/2016(H5N8)的8个基因均与2016-2017年自江苏、山东、山西、内蒙古、西藏等地区外环境中分离的H5N8 AIVs聚在一起。另外,2株分离病毒多个基因与2011-2012年自华东家鸭中分离的H5N8 AIVs的遗传距离较近。
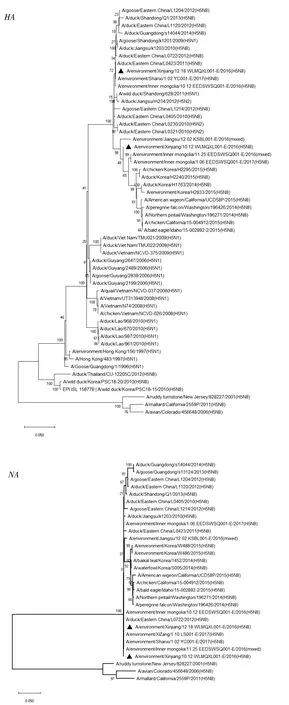
▲.本研究分离的病毒株
2.3 AIV分子特征分析2株H5N8分离株HA蛋白裂解位点处序列分别为RERRRKR↓GLF(“↓”表示裂解位点)和REKRRKR↓GLF, 均含5个连续的碱性氨基酸,为HPAIV的特征。分离病毒存在增强病毒毒力和致病力的多个突变(表2),HA发生S137A和T160A突变,可增强病毒与哺乳动物SA α2,6-Gal受体结合的亲和力[16-17],PB2的L89V、PB1的I368V、PB1-F2的N66S、NS1的P42S以及M1的N30D和T215A等突变可增强病毒对小鼠等哺乳动物的毒力和致病力均已见报道[12-15,18-20]。
表2 H5N8 AIVs分离株中与致病力相关的突变
3 讨论
新疆自2005年以来接连发生H5N1亚型高致病性AI疫情,且近年连续发生多种亚型的AI疫情,并出现人感染H7N9 AIV病例[21]。2016年开始进入频发期,尤其是2019年底至2020年初,新疆多个地区接连发生多起迁徙天鹅感染HPAIV的疫情[22]。大量的研究表明,LPM往往是多种AIV亚型共存的“大熔炉”,是人感染AIV的主要源头[6-11]。为此,对LPM中家禽和外环境定期进行AIV监测是AI防控的重要环节。本研究于2016年10月至2018年12月持续采集乌鲁木齐某大型LPM外环境样品进行AIV分离鉴定,分别自2016年冬季样品中分离到2株H5N8 AIVs,自2017年春季样品中分离到1株H9N2 AIV,说明该市LPM外环境存在AIV污染,王六合等[23]亦报道乌鲁木齐市LPM活禽和外环境中AIV阳性率较高,故尚需要加强对LPM进行定期消毒和持续监测,以降低AIV通过活禽交易传播的风险。另外,2017年4月至2018年12月采集的样品中均未分离到AIV,其与2016年底至2017年初乌鲁木齐市逐步将活禽交易集中于指定LPM并加强管理和监督有关,说明LPM每日清洗、每周休市1 d集中消毒等监管措施有效地消杀了环境中的AIV。
自2010年华东地区首次发现新的H5N8 AIV(A/duck/Jiangsu/k1203/2010)以来[1],其已快速传播至多个地区并引发禽流感疫情。据报道目前流行的H5N8 AIVs是家禽“HPAI H5Ny和HxN8”循环至野鸟时发生重配而产生[24],且H5N8 AIV可随候鸟迁徙而广泛传播[25]。本研究分离的2株环境源H5N8 AIV在序列一致性和进化关系方面存在差异,同时也存在明显的共性。2株H5N8 AIVs各基因片段中,XJ/Env7/2016(H5N8)的PB2与山东地区野鸟源H5N1 AIVs一致性最高,其余基因片段均与2016年底至2017年初自西藏、内蒙古和江苏等地区外环境中分离的H5N8 AIVs一致性最高,且部分基因与2010-2012年华东地区家鸭中分离的H5N8 AIVs一致性亦为最高。遗传进化分析表明,除XJ/Env7/2016(H5N8)的PB2与2010-2012年自华东家鸭中分离的H5N8 AIVs聚在1个分支之外,2株病毒其余各基因均与2016-2017年自江苏、山东、山西、内蒙古、西藏等地区外环境中分离的H5N8 AIVs聚在一起,同时病毒多个基因与2011-2012年自华东家鸭中分离的H5N8 AIVs的遗传距离较近。以上结果说明乌鲁木齐市LPM 环境源H5N8 AIVs与2016-2017年我国多个地区流行的H5N8 AIVs毒株进化关系密切,且多个基因源于2010-2012年华东地区家禽中流行的毒株。新疆是全球候鸟迁徙的重要通道,通过途径的“中亚―印度线”和“东非―西亚线”迁徙线与西伯利亚、蒙古国、印度半岛以及国内多个地区连接,AIVs随候鸟迁飞传入新疆的风险较高,前期我们在该区域野鸟粪便中分离到多种亚型AIVs[26],近几年新疆多个地区接连发生多起迁徙候鸟感染AIVs的疫情[22]。本研究中XJ/Env7/2016(H5N8)的PB2与野鸭源H5N1 AIVs病毒株遗传距离较近,2株分离病毒的HA、NA、PA也与多个野鸟源病毒株的遗传距离较近,说明分离病毒的一些基因源于野鸟,且2016年底和2017年初多个地区分离的环境源H5N8 AIVs间遗传距离较近,说明这些毒株的分布地域极为广泛,可能是通过迁徙候鸟的跨地域快速传播所引起的。
2株分离病毒的HA裂解位点均含5个连续的碱性氨基酸,为HPAIV;受体结合位点226,228位未发生突变,保留为Q和G,倾向于结合禽类α2,3半乳糖苷唾液酸(SAα2,3-Gal)受体,而S137A和T160A突变可增强AIVs与哺乳动物SAα2,6-Gal受体的结合[16-17],T160A常发生于感染人的HPAIVs[27],可增强AIVs在豚鼠和雪貂等哺乳动物中的传播性[17]。除HA之外,分离毒株还存在多个突变(表2)可能影响病毒致病性和感染宿主特异性等方面的特性,如PB2的L89V[12-13]和PB1-F2的N66S[15]可提高病毒在哺乳动物细胞中的复制活性和对小鼠的毒力,NS1的 P42S[20]以及M1的N30D和T215A[18]突变均可增强AIVs在小鼠中的毒力, Env-XJ-1-H5N8 PB1的I368V突变可增强H5亚型AIVs的传播性[14],上述突变中的一些也存在于野鸟源H5N8 AIV[28-29]和人感染的HPAI-Vs[30]。
本研究从新疆乌鲁木齐市LPM外环境中分离的2株H5N8 AIVs呈高致病性,其与2016-2017年分离自国内多个地区外环境中的H5N8 AIVs的遗传关系较近,可能随野鸟迁飞而广泛传播,另外,分离毒株多个基因与2010-2012年华东地区家鸭中分离的H5N8 AIVs的遗传距离接近,其多个突变可增强病毒的致病性和跨物种传播的能力,有潜在引发AI疫情和威胁公共卫生的风险。
