血清1型禽腺病毒JS2017株感染性克隆构建及病毒拯救
2022-01-25张小荣王延龙郭梦娇张成成薄宗义曹永忠吴艳涛
张小荣,王延龙,郭梦娇,张成成,薄宗义,曹永忠,吴艳涛,*
(1.扬州大学 兽医学院 江苏省动物重要疫病预防控制协同创新中心,江苏 扬州 225009; 2.扬州大学 农业科技发展研究院 教育部农业与农产品安全国际合作联合实验室,江苏 扬州 225009)
禽腺病毒(fowl adenovirus,FAdV)根据群抗原特异性差异可以分为3个群[1]。Ⅰ群禽腺病毒最为常见,临床上主要引起包涵体肝炎和心包积液综合征,分为12个血清型,代表株为鸡胚致死性孤儿病毒(CELOV);Ⅱ群禽腺病毒包括火鸡出血性肠炎病毒、雉鸡大理石脾病病毒及鸡大脾病病毒;Ⅲ群禽腺病毒包括减蛋综合征病毒及来自鸭的相似病毒。
腺病毒载体是目前研究和应用最广的载体系统之一,其中以人源腺病毒载体的研究最为广泛,已经有相关产品进入商业化应用。近年来,随着对禽腺病毒研究的逐渐深入,以禽腺病毒为载体开发禽用基因工程疫苗的研究也相继被报道,常用作载体研究的病毒包括Ⅰ群禽腺病毒中的血清1、4和9型(FAdV-1、FAdV-4、FAdV-9),展示出良好的应用前景[2-5]。
近年来,腺病毒的反向遗传学操作技术已经比较成熟,该技术通过将腺病毒全长基因组DNA克隆入质粒载体获得感染性克隆,可根据需要在DNA水平对病毒基因组进行遗传操作,然后将感染性克隆转染细胞即可直接获得重组病毒[4,6]。
本研究的目的是以实验室前期分离和鉴定的1株天然无致病性的FAdV-1(JS2017株)为研究对象,将其完整基因组DNA克隆到黏粒载体内,构建感染性克隆,成功建立FAdV-1反向遗传操作技术平台,为进一步将FAdV-1作为活病毒载体研发禽用基因工程疫苗提供平台。
1 材料与方法
1.1 毒株、细胞株血清1型禽腺病毒(FAdV-1)JS2017株由本实验室分离、鉴定和保存[7-8];鸡肝癌细胞LMH购自美国模式菌种收集中心(ATCC),编号为CRL-2117。
1.2 质粒与菌株pCR2.1载体购自Invitrogen公司;黏粒载体SuperCos-1及其配套试剂购自Agilent公司;pEASYTM-T3克隆载体及Trans-T1感受态细胞购自北京全式金生物科技有限公司;大肠杆菌DH10B由本实验室保存。
1.3 主要试剂dNTPs、EasyTaq DNA polymerase(5 U/μL)、T4DNA连接酶购自北京全式金生物技术有限公司;LipofectamineTM3000转染试剂购自Thermo Fisher Scientific公司;限制性内切酶均购自New England Biolabs(NEB)公司;质粒小量提取试剂盒与DNA凝胶回收试剂盒均购自Axygen公司;DNA Marker购自宝生物工程(大连)有限公司;其他普通生化试剂均为国产分析纯试剂。
1.4 FAdV-1 JS2017株病毒基因组DNA提取将FAdV-1 JS2017株病毒按MOI=0.1接种单层LMH细胞,培养4~5 d,待出现明显细胞病变,同时收取细胞与培养上清,反复冻融3次后10 000×g离心10 min 后取上清经超滤管10倍浓缩。浓缩液中加入等体积裂解液(10 mmol/L Tris-HCl(pH 8.0)、10 mmol/L EDTA、1% SDS、100 mg/L蛋白酶K),37℃反应13 h后经酚∶氯仿∶异戊醇(25∶24∶1)抽提,乙醇沉淀回收DNA,TE溶液(pH 8.0)溶解沉淀,测定浓度后4℃保存备用。
1.5 UP、DOWN同源臂的扩增与克隆根据JS2017株的全基因组序列(GenBank登录号:MK050972),选取FAdV-1基因组左端1 186 bp(1~1 186 nt)、右端1 130 bp(42 610~43 739 nt)作为UP、DOWN同源臂,设计2对扩增引物FA-UF/R、FA-DF/R,分别用于UP、DOWN同源臂的扩增。在UP同源臂的上游引物5′端引入BamHⅠ和AscⅠ酶切位点,下游引物5′端引入SbfⅠ和NotⅠ酶切位点。在DOWN同源臂上游引物5′端引入SbfⅠ酶切位点,下游引物5′端引入 ApaⅠ、BamHⅠ和AscⅠ酶切位点。引物序列见表1,由南京金斯瑞生物科技有限公司合成。以病毒全基因组DNA为模板,PCR扩增同源臂。回收、纯化同源臂扩增产物,并克隆进pEASYTM-T3克隆载体内,转化入Trans-T1感受态细胞,按说明书方法通过PCR筛选阳性克隆进一步进行测序验证,重组克隆分别命名为pT-U和pT-D。
1.6 质粒pCos-UD的构建
1.6.1转移质粒pCR-UD的构建 利用BamHⅠ和NotⅠ双酶切质粒pT-U和pCR2.1,回收UP同源臂和线性化载体pCR2.1,经T4DNA连接酶连接,获得质粒pCR-U。随后用ApaⅠ和SbfⅠ双酶切质粒pT-D和pCR-U,回收DOWN同源臂和pCR-U载体片段,经T4DNA连接酶连接,构建质粒pCR-UD,并利用BamHⅠ酶切进行鉴定。
1.6.2质粒pCos-UD的构建 黏粒载体SuperCos-1中含有新霉素抗性基因,与设计方案后续步骤中抗性基因的使用相冲突,因此首先利用Hind Ⅲ和SmaⅠ限制性内切酶对SuperCos-1进行双酶切去除新霉素抗性基因,通过Klenow大片段对DNA末端补平后重新进行连接获得改造后质粒SuperCosΔner。BamHⅠ分别酶切质粒SuperCos-Δner、pCR-UD,回收后,经T4DNA连接酶连接同源臂UD与线性化质粒SuperCosΔner,构建质粒pCos-UD。
1.7 感染性克隆pCos-FAdV1的构建首先,利用SbfⅠ酶切线性化质粒pCos-UD,回收酶切产物。取线性化pCos-UD与1.4中提取的病毒全基因组DNA共同电转化到BJ5183感受态细胞内,利用宿主菌BJ5183所携带的重组酶催化病毒基因组DNA末端与线性化pCos-UD中携带的UP和DOWN同源臂发生同源重组,从而将完整病毒基因组DNA整合入载体中。电转化条件为电压2 500 V、电阻200 Ω、电容25 μF,转化后的细菌于37℃培养复苏1 h后,均匀地涂布在含有100 mg/L氨苄青霉素的LB平板上,过夜培养,挑取单菌落,使用禽Ⅰ群腺病毒通用引物Hex-F/R(表1)进行重组克隆筛选[1]。将筛选出的阳性感染性克隆命名为pCos-FAdV1。感染性克隆构建流程如图1所示。由于大肠杆菌BJ5183内部含有重组酶,有可能会引起感染性克隆中FAdV1基因组中重复序列的重组,造成基因重排,为防止这种现象发生,将在BJ5183内重组获得的感染性克隆pCos-FAdV1重新电转化入缺少重组酶基因的大肠杆菌DH10B中。
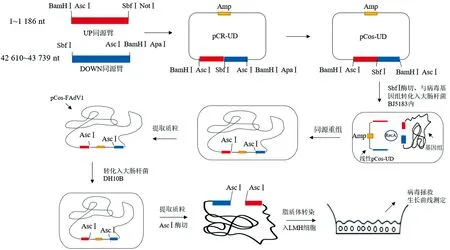
图1 感染性克隆构建流程图
1.8 感染性克隆pCos-FAdV1的鉴定采用2种方法分别对感染性克隆pCos-FAdV1进行鉴定。首先针对感染性克隆不同位置共设计5对引物:F-fiber1/2、F-orf10、F-orf11、FCS1、FCS2(表1),通过PCR进行鉴定。然后取PCR鉴定正确的单克隆进行扩大培养,提取质粒,利用限制性内切酶Hind Ⅲ对感染性克隆进行酶切图谱分析。

表1 感染性克隆pCos-FAdV1构建及鉴定引物
1.9 病毒拯救及复制能力的测定
1.9.1病毒cFAdV-1的获取 利用在同源臂末端引入的限制性内切酶AscⅠ酶切感染性克隆pCos-FAdV1,释放完整的FAdV1基因组DNA片段,通过酚∶氯仿抽提和乙醇沉淀法回收酶切产物。按照LipofectamineTM3000转染试剂说明书,将酶切产物转染进LMH细胞,37℃培养3~4 d后,收取细胞及上清,反复冻融后取100 μL,进行盲传,直至出现明显的细胞病变,收取细胞与上清,反复冻融后,继续接种LMH细胞进行扩大培养。将拯救出的病毒命名为cFAdV-1。
1.9.2生长曲线的测定 取病毒cFAdV-1,以及母本病毒JS2017株,使用无血清DMEM培养基稀释后按照MOI=0.1接种到状态良好的LMH单层细胞,37℃培养箱吸附2 h后,更换为含有2% FBS的DMEM维持液。取接种后0,12,24,36,48,60,72 h的细胞与上清,反复冻融后进行TCID50的测定,根据测定结果绘制病毒生长曲线,每个时间点设置3个重复。
2 结果
2.1 FAdV-1 JS2017株病毒全基因组的提取提取的病毒基因组经分光光度法进行测定,质量浓度为700 mg/L,D260/D280=1.90。
2.2 UP、DOWN同源臂的扩增以提取的病毒全基因组为模板,采用同源臂扩增引物FA-UF/R、FA-DF/R对基因组1~1 186,42 610~43 739 nt 2个片段进行扩增,扩增产物经琼脂糖凝胶电泳后,大小分别为1 186,1 130 bp,与预期大小一致(图2)。
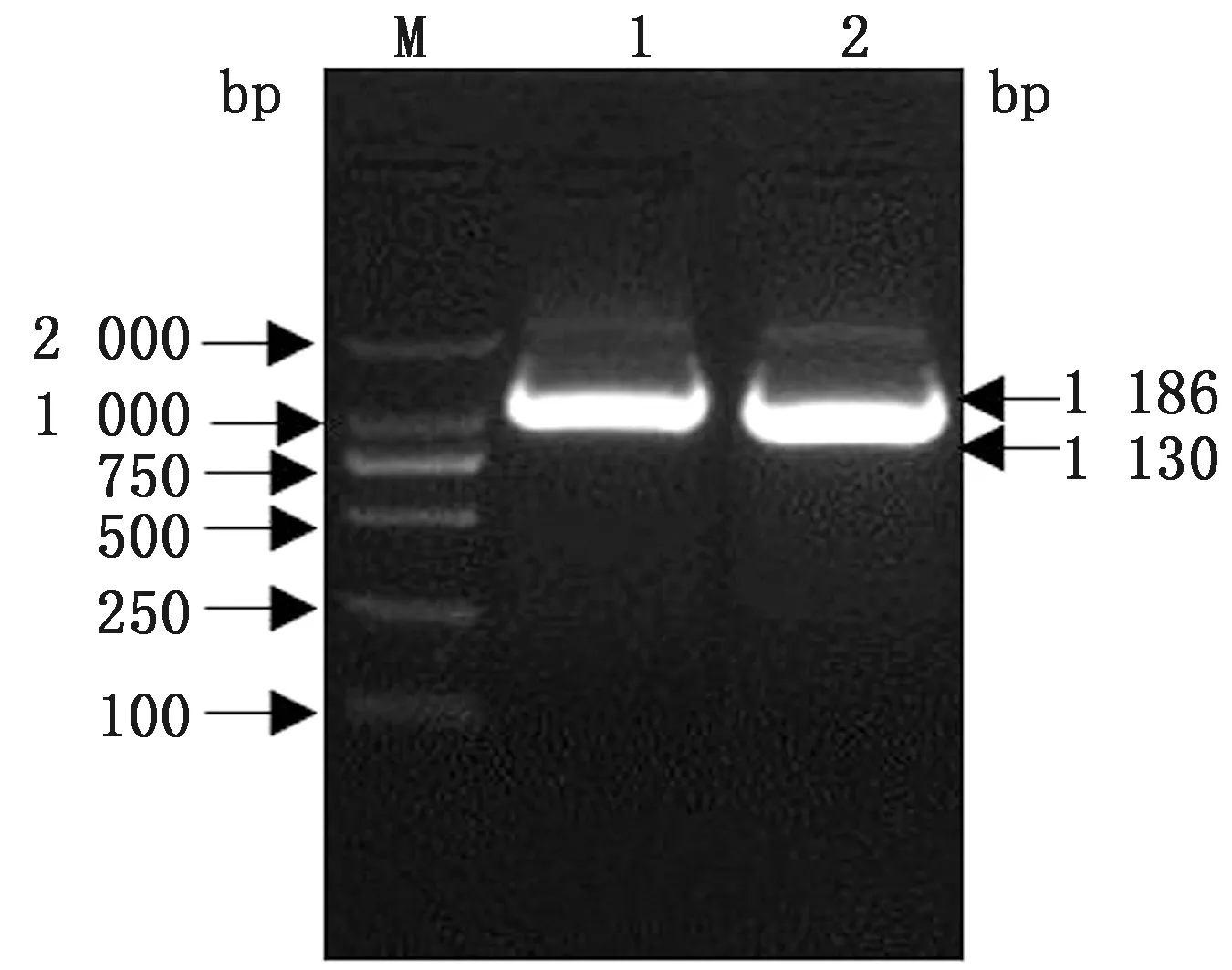
M.DL2000 DNA Marker;1.UP同源臂扩增产物;2.DOWN同源臂扩增产物
2.3 质粒pCos-UD的构建
2.3.1中间质粒pCR-UD的构建与鉴定 将UP、DOWN同源臂克隆进质粒pCR2.1,构建中间质粒pCR-UD,经BamHⅠ酶切后电泳可见4 000,2 316 bp 大小的条带,与预期一致(图3)。
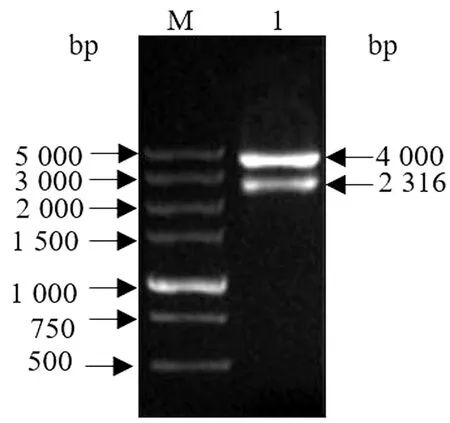
M.DL5000 DNA Marker;1.质粒pCR-UD酶切产物
2.3.2质粒SuperCos-Δner的构建与鉴定 删除质粒SuperCos-1的新霉素抗性基因,获得质粒SuperCos-Δner。经XbaⅠ和BamHⅠ双酶切后电泳可见条带大小分别5 481,1 140 bp,而原始质粒Supercos-1酶切后产生的条带大小分别为6 800,1 140 bp,鉴定结果如图4 所示,说明新霉素抗性基因已被成功删除。
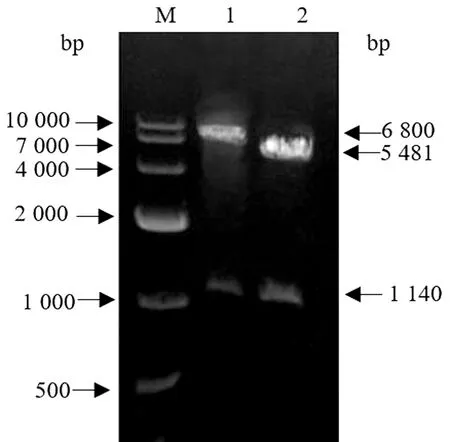
M.DL10000 DNA Marker;1.SuperCos-1酶切产物;2.SuperCos-Δner酶切产物
2.3.3质粒pCos-UD的鉴定 将同源臂UP和DOWN整体克隆进SuperCos-Δner获得质粒pCos-UD。利用限制酶EcoRⅤ和XbaⅠ对质粒pCos-UD进行双酶切鉴定,电泳结果显示目的条带大小为5 068,3 869 bp,与预期一致(图5)。
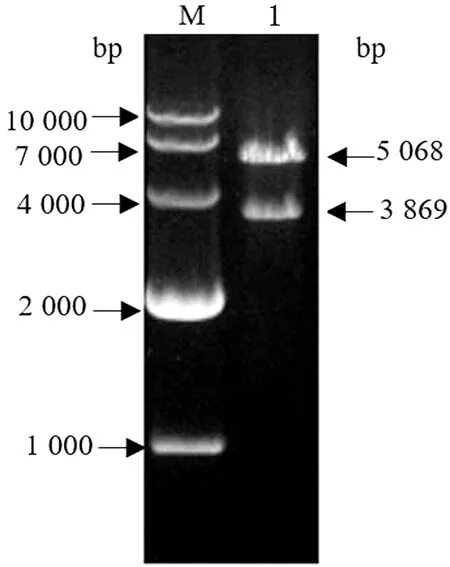
M.DL10000 DNA Marker;1.pCos-UD酶切产物
2.4 感染性克隆pCos-FAdV1的筛选和鉴定将线性化质粒pCos-UD与FAdV-1病毒全基因组DNA共转化入大肠杆菌BJ5183内,通过同源重组的方法获得包含完整病毒基因组的感染性克隆,经FAdV1通用检测引物Hex-F/R 扩增产物大小约为800 bp(图6)。
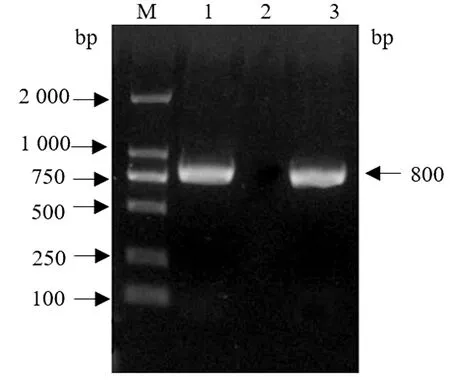
M.DL2000 DNA Marker;1.阳性感染性克隆;2.阴性对照;3.FAdV-1 DNA
将经通用引物PCR筛选到的感染性克隆转化入大肠杆菌DH10B内,分别以5对引物进行PCR鉴定,PCR产物电泳结果如图7所示,引物F-fiber1/2、F-orf10、F-orf11、FCS1和FCS2扩增产物大小分别为801,911,468,1 085,898 bp,鉴定结果均与预期一致。以Hind Ⅲ对感染性克隆进行酶切图谱分析,电泳结果见图8,感染性克隆酶切后产生的条带大小为12 250,9 193,6 845,5 446,4 748,4 108,4 046,3 736 bp,与预期一致。
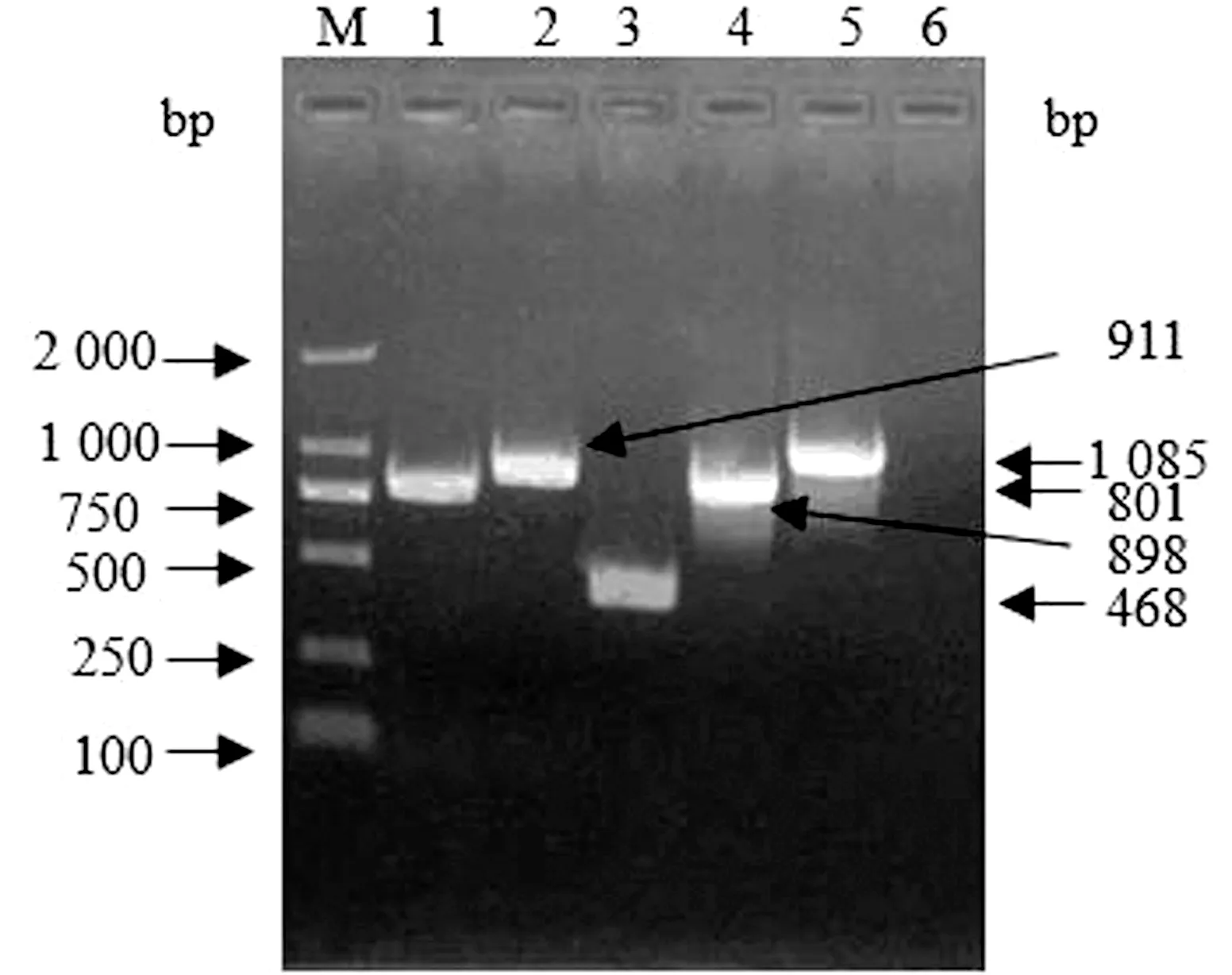
M.DL2000 DNA Marker;1.引物F-fiber1/2;2.引物F-orf10;3.引物F-orf11;4.引物FCS2;5.FCS1;6.阴性对照
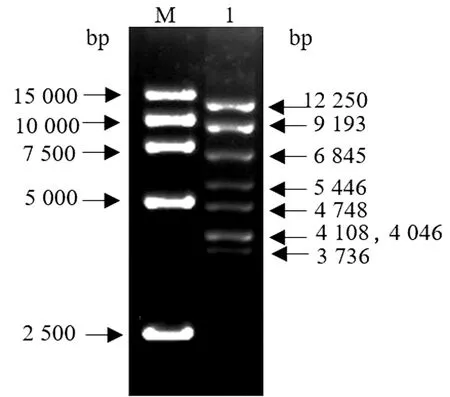
M.DL15000 DNA Marker;1.感染性克隆 pCos-FAdV1酶切产物
2.5 病毒拯救结果将AscⅠ酶切从感染性克隆pCos-FAdV1中释放出的FAdV1基因组DNA转染入LMH细胞并进行盲传,在盲传第2代时观察到FAdV感染细胞产生的典型细胞病变(图9),细胞间距增加、变圆、折光性增强。经生长曲线测定,拯救出的病毒cFAdV-1与亲本病毒JS2017株生长趋势基本一致,说明具有相似的复制能力(图10)。
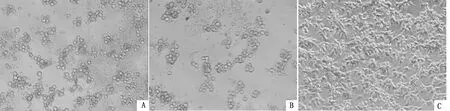
A.接种cFAdV-1的LMH细胞;B.接种FAdV-1(JS2017)的LMH细胞;C.未接种病毒的LMH细胞
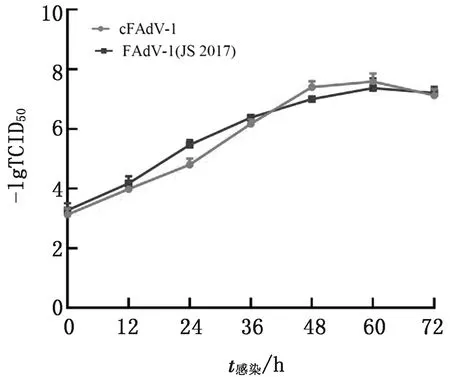
图10 病毒cFAdV-1与FAdV-1(JS2017)生长曲线测定结果
3 讨论
腺病毒载体是最具应用前景的病毒载体之一,研究最广泛的是人腺病毒载体,已有部分产品进入临床应用,其中血清型5型人腺病毒被用于新冠肺炎疫苗的研发[9]。禽腺病毒载体在基因工程疫苗研发方面同样展现出良好的应用前景,但是国内对于禽腺病毒载体的研究起步较晚。
重组腺病毒常用的构建方法是在真核细胞中将2个重叠的病毒DNA片段之间发生同源重组,从中筛选出含有重组病毒的空斑。此方法同源重组效率低,并且需要对重组病毒进行多轮费时、费力的纯化过程。而CHARTIER等[10]基于相同的原理,在大肠杆菌中完成重组病毒的构建,细菌内同源重组法不仅克服了真核细胞内同源重组效率低的缺点,且可以直接利用抗性筛选含有重组腺病毒载体的大肠杆菌,后期无需进行繁琐的病毒纯化过程。禽腺病毒感染性克隆常在含有RecA重组酶的大肠杆菌BJ5183内构建,可以直接用于重组病毒的构建,后期只需线性化重组感染性克隆并转染入合适的宿主细胞内,便可以获得纯的重组病毒,无需病毒纯化过程。FRANCOIS等[11-12]首次在黏粒载体中构建含禽腺病毒全基因组的感染性克隆,通过基因突变对病毒复制非必需区进行了验证,构建了表达传染性法氏囊病毒VP2蛋白的重组禽腺病毒。最近该方法又被成功用于FAdV-4感染性克隆的构建[4]。
本研究同样基于同源重组的原理,首先将FAdV-1 JS2017株基因组左右末端的序列顺序插入黏粒载体SuperCos-1中,再与从病毒中提取的完整基因组DNA共转化大肠杆菌BJ5183,利用BJ5183宿主菌体内带有的重组酶催化病毒末端同源区之间的重组,从而将JS2017株病毒基因组完整插入质粒载体中。为鉴定克隆到载体中的病毒基因组完整性,首先针对病毒基因组与载体的连接区和病毒基因组不同部位设计了5对引物进行PCR初步验证,然后选择合适的酶切位点进行切割,分析其酶切图谱,最后转染LMH细胞进行病毒拯救,结果均证明JS2017株基因组已经按照预期设计完整插入质粒载体中,感染性克隆构建取得了成功,且拯救出的病毒与亲本毒株在生物学特性方面无明显差异。
综上所述,本研究构建的FAdV-1感染性克隆,可为今后以FAdV-1为载体开展基因工程疫苗的研究提供平台,同时也可以用于探索FAdV-1基因组中未知功能的ORF在病毒复制及其与宿主相互作用中的地位。
