Detection of pigeon circoviruses in ticks of sheep and camels in Inner Mongolia,China
2022-01-25KONGYunyiYANChaoZHANGGangCAIYurongHEBiaoLIYong
KONG Yunyi,YAN Chao,ZHANG Gang,CAI Yurong,HE Biao,LI Yong*
(1.Key Laboratory of Ministry of Education for Protection and Utilization of Special Biological Resources in the Western China,Ningxia University,Yinchuan 750021,China;2.School of Life Science,Ningxia University,Yinchuan 750021,China;3.Key Laboratory of Jilin Province for Zoonosis Prevention and Control,Institute of Military Veterinary Medicine,Academy of Military Medical Sciences,Academy of Military Sciences,Changchun 130122,China)
Abstract:Circovirus is one of the smallest known DNA viruses known to be able to infect animals.In this report,circovirus was detected by high-throughput sequencing in two tick species,Hyalomma asiaticum and Dermacentor nuttalli collected from sheep and camels in Inner Mongolia,China.A complete genomic sequence of 2 042 base pairs of cyclic single-stranded DNA(ssDNA) was obtained by reverse semi-nested PCR amplification.Evolutionary analysis further revealed that it belonged to circovirus with two major open read frames(ORFs) encoding replicase and capsid proteins,respectively.Sequence and phylogenetic analysis suggested that the circovirus belonged to pigeon circovirus.The results demonstrated the presence of pigeon circovirus in ticks in Inner Mongolia,which provide the basis for future exploration of the prevalence of pigeon circovirus in Inner Mongolia and the pathogens carried by ticks in Inner Mongolia.
Keywords:circovirus;pigeon circovirus;ticks;phylogenetic analysis
Taxonomically,circovirus is a small,nonenveloped,single-stranded DNA virus with a circular genome,which is categorized into the genusCircovirusof theCircoviridaefamily[1].Circoviral genomes range from 1.8 to 2.1 kb in size and contain two open reading frames that encode the replication(Rep) and capsid (Cap)proteins[2].Vertebrates,including birds,poultry and mammals,are natural hosts of circovirus.Circovirus infection may cause diseases in animals[3].For example,porcine circovirus type 2(PCV2) is associated with various disease syndromes in pigs,primarily postweaning multisystemic wasting syndrome(PMWS)[4].Other symptoms in sows include abortion,dermatitis,and nephropathy syndrome[5-7].Canine circovirus infection has also been reported to cause hemorrhagic enteritis that was associated with a sudden onset of weakened appetite,vomiting,and bloody diarrhea in dogs[8-9].Similarly,avian circoviruses are related to symptoms of immunosuppression and feather abnormalities in birds or poultry[10-13].
Although porcine circovirus(PCV) is the only circovirus capable of being culturedinvitroin PK-15 cells[14],an increasing number of novel circovirus-like genomes have been discovered in wild mammals and insects using high-throughput sequencing techniques[15-16].However,although porcine circovirus and raven-related circovirus have been found in ticks,there are few reports of circovirus in ticks,which are the second most common vectors of pathogens[17-18].To explore whether the ticks in Inner Mongolia Autonomous Region were the vectors of circovirus,we tried to detect circular ssDNA genomic sequences from ticks collected from sheep and camels in this location by high-throughput sequencing technology and the reverse semi-nested PCR.
1 Materials and methods
A total of 2 662 ticks were collected from sheep and camels in the Alxa Left Banner,Alxa Right Banner and Siziwang Banners of Inner Mongolia from April to May 2016.For each sampling site,three herds of sheep or camels were selected,with more than 50 animals in each herd were randomly selected for sampling.Ticks on the skin surface of animals were caught and collected in a container.All samples were frozen and stored at -80℃.The collected ticks were pooled according to their hosts from which they were collected and the species of ticks.Approximately 15 ticks were pooled in one sample for processing and analysis.Hyalommaasiaticumwas mainly distributed in Alxa Left Banner,andDermacentornuttalliwas mainly distributed in Siziwang Banner,although both were found in Alxa Right Banner.Among the collected tick samples,2 388Hyalommaasiaticumticks were separately pooled into 160 samples,and the other 274Dermacentornuttalliticks were pooled into 18 samples(Table 1).
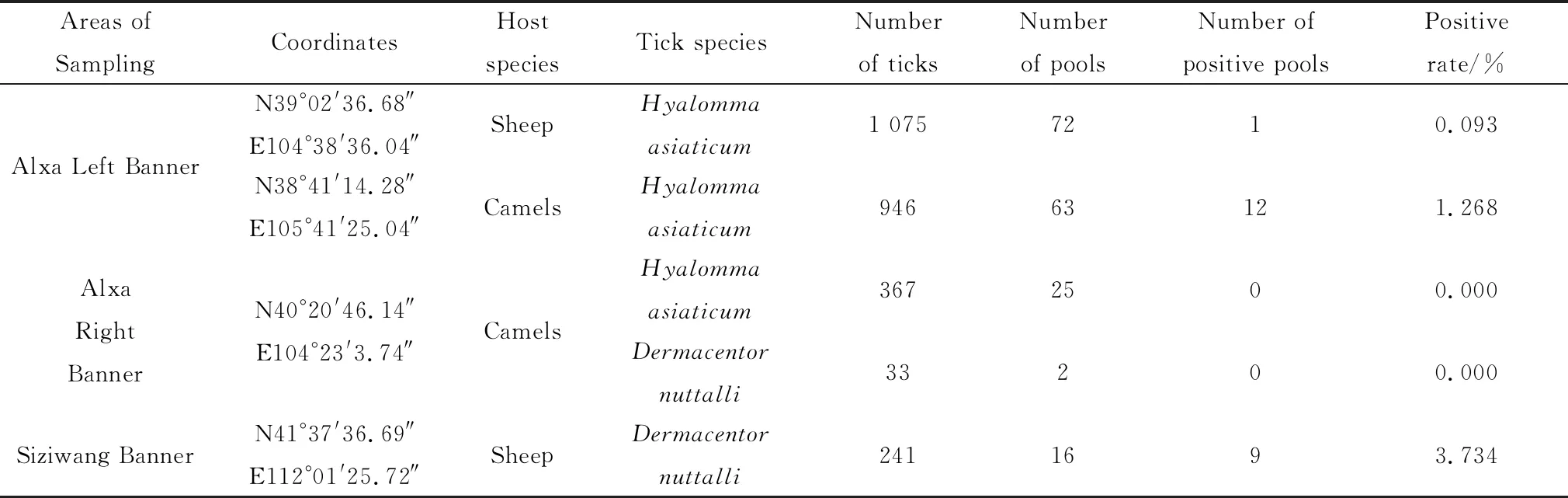
Table 1 Molecular detection of circovirus from ticks isolated from different regions in Inner Mongolia,China
To prepare samples,ticks of each pool were put into a 2 mL centrifuge tube containing 1 mL of Dulbecco’s modified Eagle’s medium(DMEM;Gibco,Carlsbad,USA) prior to being homogenized in a frozen grinder(Jingxin,Shanghai,China).The homogenized samples were then centrifuged at 4℃ and 12 000×gfor 10 min.The supernatants were collected,and total RNA was extracted from tick pools with RNeasy Mini Kit(QIAGEN,Hiden,Germany),and then the cDNA was synthesized using M-MLV reverse transcriptase(TaKaRa Biotechnology,Dalian,China).The qualified cDNAs were subjected to high-throughput sequencing for viral metagenomic analysis by the Beijing Genome Institute(BGI,Shenzhen,China).
After removing host-derived sequences,the 3 568 561 reads with an average length of 145 nt were obtained,of which 12 931(0.004%) were annotated to viruses of 20 families and some unclassified viruses,including dsDNA,ssDNA,and ssRNA viruses.According to the sequencing results,the DNA of each tick pool was extracted using a TIANamp Genomic DNA Kit(Tiangen,Beijing,China),and the fractional genes of circovirus were determined by PCR using the primers TiCV-F1,TiCV-F2 and TiCV-R(Supplemental Table 1).The viral metagenomic results were also corroborated by a PCR assay.To obtain the complete sequence of circovirus,primers for amplifying the complete genome were designed according to the results of high-throughput sequencing(Supplemental Table 2).The PCR was performed in a 50 μL reaction system containing 1 μL of genomic DNA as a template from each pool sample,25 μL of 2×Phabta Max Master Mix(Vazyme,Nanjing,China),20 μL of ddH2O,and 2 μL each of the forward and reverse primers (10 μmol/L).The PCRs were conducted in a thermal cycler(Analytik Jena,Thuringia,Germany) using the following thermocycling conditions:predenaturation at 95℃ for 3 min,followed by 35 cycles(denaturation for 15 s at 95℃,annealing for 15 s at 57℃,and extension for 15-47 s at 72℃),and a final incubation for 5 min at 72℃.The PCR products were analyzed by electrophoresis using a 1.0% agarose gel stained with ethidium bromide.
All amplified products were analyzed by Sanger sequencing(ComateBio,Changchun,China).Nucleotide sequence homology was analyzed by BLAST(http://www.ncbi.nlmn.nih.gov/BLAST).All the sequencing results were assembled and aligned using CLUSTAL W in MEGA 7.0(http://www.megasoftware.net/) and compared with the sequences of other circovirus strains in GenBank.A phylogenetic tree was constructed with the maximum-likelihood method by MEGA 7.0 software.The reliability of the branches of the tree was assessed using a bootstrap analysis with 1 000 replicates.
2 Results
All samples were screened for the presence of circovirus by half-nested PCR.The distribution of circovirus in these three regions was assessed,and circoviral DNA was detected in tick samples fromAlxa Left Banner and Siziwang Banner but not in samples from Alxa Right Banner.The positive percentages of circoviral DNA were 0.093% in ticks from sheep and 1.268% in ticks from camels in Alxa Left Banner,Inner Mongolia and 3.734% in ticks from sheep in Siziwang Banner,Inner Mongolia.The positive rate was determined by calculating the minimum infection rate(MIR),which was the ratio of the number of positive tick pools to the total number of analyzed ticks[19].The analysis data were listed in Table 1.
The complete sequences of circoviruses amplified by semi-nested PCR using the specific primer sets based on Sanger sequencing revealed a circular viral genome of 2 042 nucleotides(nt) in ticks.The genomic sequence obtained in this study was submitted to GenBank with accession number MN920392.
To explore the evolutionary relationship between this circovirus and other circoviruses,the complete genomic sequences of circoviruses were used for phylogenetic tree mapping and homology analysis.Genome sequences of 19 members of this family were analyzed.Phylogenetic analysis of the complete viral genome showed that this virus was close to pigeon circovirus(PiCV)(Fig.1A).In addition,phylogeny also suggested that tick circovirus(TiCV) had a common ancestor with the evolutionary branches of starling circovirus(StCV),raven circovirus(RaCV),finch circovirus(FiCV) and zebra finch circovirus.Except for chimpanzee feces-associated circovirus(accession no.GQ404851),mammalian and avian circoviruses were relatively independent of the evolutionary tree.
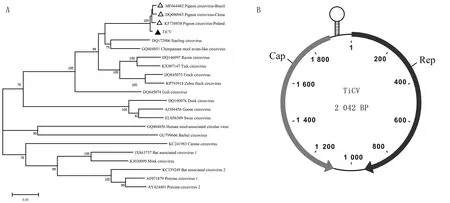
A.Phylogenetic tree of the tick circovirus(TiCV) complete sequence.The tree was constructed using the maximum-likelihood method with MEGA 7.0 software.Bootstrap testing(1 000 replicates) was performed,and values lower than 60% were not shown.The bar at the bottom of the figure denotes the distance;B.Genome map of TiCV showing the ORFs encoding Rep and Cap
Alignment analysis of the TiCV genome with those of other circoviruses revealed a high sequence identity of 95.9% to 95.6% with the genome of PiCV and a low identity(43.9% to 61.1%) with duck circovirus(DuCV),goose circovirus(GoCV),gull circovirus(GuCV),RaCV,FiCV and StCV.There were two large ORFs for this circovirus,which encode the Rep and Cap.The intergenic region between the two ORFs could form a potential stem-loop structure,according to the mfold web server(Fig.1 B).The putative Rep protein of TiCV was 315 amino acids(aa),and had a high identity with the Rep proteins of FiCV(76.5%),RaCV (76.5%),StCV(76.9%),and PiCVs that were isolated from China(98.1%) and Brazil(97.2%).It had a low identity with those of GuCV(64.9%),human stool-associated circovirus(54.9%),DuCV(54.3%),and GoCV(51.9%)(Table 2).The putative Cap protein of TiCV was 273 aa and shared a higher identity with that of PiCVs from China (99.2%) and Brazil(98.8%) than that of with other circoviruses,including FiCV(54.5%),GuCV(56.4%),and StCV(66.1%)(Table 2).

Table 2 Comparison of amino acid and nucleotide sequence identity between tick circovirus and other circoviruses
3 Discussion
Ticks are efficient vectors for the transmission of pathogens,including many arboviruses.To our knowledge,the identified tick-borne viruses include members of two orders,nine families and at least twelve genera[20].The infection of most of these tick-borne viruses may cause diseases in humans or animals[21].With a vast territory and well-balanced ecological environment in Inner Mongolia,it relies heavily on the animal husbandry industry.In this report,we attempted to detect circular ssDNA genomic sequences from ticks collected from sheep and camels in Inner Mongolia,China,by high-throughput sequencing technology.
Notably,genomic DNA of PiCV was detected in ticks parasitized on sheep and camels at two remote sampling sites in Inner Mongolia,suggesting that PiCV exists in ticks in Inner Mongolia.Interestingly,however,the ticks were collected from sheep and camels herding on the desert steppe or meadow grassland in Inner Mongolia,where there is no pigeons live.
Inaddition,it has been reported that a PiCV was detected in chicken and that a PCV was detected in cow or calf samples[22-23],suggesting that circovirus may transmit between different species.However,there is no direct evidence that this virus can infect ruminants,such as sheep or camels.These findings suggest that the serological detection method using antibodies against circovirus are needed in further research in addition to nucleic acid detection methods.Notably,the role that ticks play in the transmission route of circovirus is currently uncertain.Whether ticks are used as a source of transmission to spread the virus to the host or as an intermediate host from an infected animal to a tick is worthy of additional investigation for further validation by serological detection[24].
