花生基因组学在遗传育种中的研究进展
2022-01-24陈小平洪彦彬李少雄梁炫强
陈小平,鲁 清,洪彦彬,李少雄,梁炫强
(广东省农业科学院作物研究所/广东省农作物遗传改良重点实验室,广东 广州 510640)
花生(Arachis hypogaeaL.)是我国重要的油料经济作物,是食用植物油和食用蛋白的重要来源之一。花生营养价值高,含有丰富的油脂(35%~60%)和蛋白质(22%~35%)以及膳食纤维、矿物质、维生素和生物活性大分子,是非洲贫困地区人们所需营养和能量的重要来源[1]。花生起源于南美洲的阿根廷北部至玻利维亚南部区域[2-3],目前已在100 多个国家种植,主要分布在亚洲、非洲和南美洲的发展中国家。联合国粮农组织数据库(FAOSTAT)显示,2019 年全球花生种植面积达2 960 万hm2,总产量4 876 万t,其中亚洲和非洲花生种植面积占全球95%、产量占90%以上。随着花生遗传学、基因组学、育种学的发展以及耕作栽培技术的不断提升,全球花生平均单产从1961 年的849 kg/hm2提高到2019年的1 647 kg/hm2。我国是世界最大的花生生产国,总产占世界花生产量的36%,花生总产量和出口量远大于其他油料作物,是少数具有国际竞争力的出口农产品。
过去60 年我国花生产业获得显著发展,花生良种选育与推广贡献巨大,选育了一批在产量、品质(高油、高油酸、高蛋白)、抗性等方面具有优良特性的花生新品种并进行大面积推广应用[4-6]。育种技术取得长足发展,多种技术并存,常规育种与分子育种技术不断融合。随着DNA 测序技术的快速发展,推动基因组学研究日新月异,以全基因组选择(Genomic Selection,GS)为代表的多组学育种技术研究推动作物育种理论和技术的重大变革。近年来花生野生种和栽培种的参考基因组相继发布[7-12],为花生基因组学研究奠定了重要基础,进一步加快花生基因组学在遗传育种中的应用。本文综述了花生基因组进化、全基因组测序、分子标记开发、遗传图谱构建及重要性状QTL 定位、花生传统育种技术及其局限性、分子辅助育种技术及全基因组选择等基因组学和育种理论与技术的研究进展。
1 花生基因组学研究进展
1.1 花生基因组进化
栽培花生是豆科花生属(Arachis)中唯一驯化的物种。豆科花生属共有80 多个种,但只有2 个四倍体(2n=4x=40),分别为栽培种A.hypogaea和野生种A.monticola),其他均为二倍体野生种(2n=2x=20)。染色体组型分析表明,花生属物种包括A、B、D、F、K、G 等6 个不同基因组[13-15]。大部分二倍体花生属物种是A或B 基因组(AA 或BB)。细胞学研究表明,花生栽培种是异源四倍体(AABB,2n=4x=40),由2 个二倍体野生种经过一次自然杂交后染色体加倍而成[13,16],其中,A亚基因组的供体野生祖先种是A.duranensis(AA,2n=2x=20),B 亚基因组的供体野生祖先种是A.ipaensis(BB,2n=2x=20)。近年来花生基因组测序研究发现,异源四倍体花生的形成比细胞学研究推测的过程更加复杂。基因组进化分析表明,栽培花生的B亚基因组来自A.ipaensis,而A 亚基因组不只是来自A.duranensis,可能有多个来自于不同二倍体野生种的A 基因组供体。因此,花生四倍体的形成有两种可能,一是A.ipaensis与A.duranensis经过多轮杂交后两个亚基因组发生不对称进化;另一种是A.ipaensis与包括A.duranensis在内的多个A 基因组的二倍体野生种杂交最后形成四倍体栽培种[17-18]。随着基因组学发展,将有更多的花生属物种完成测序,可以进一步揭示花生属物种基因组之间的进化关系。
1.2 花生全基因组测序
近年花生基因组学蓬勃发展,参考基因组从无到有,目前已有4 个花生物种完成了全基因组测序,共产生了8 个参考基因组,包括栽培种3个不同基因型的参考基因组,极大丰富了花生基因组数据资源,使花生从基因组资源匮乏作物一跃成为基因组资源丰富作物。
花生属物种的基因组相对较大,而且重复序列多,属于复杂基因组。细胞学分析表明,二倍体花生的2C 值在2.55~3.22 pg 之间,二倍体基因组平均大小约1.4 Gb,四倍体花生的2C 值约5.7 pg,基因组大小约2.8 Gb[19]。鉴于四倍体基因组较大,花生全基因组测序研究首先从二倍体野生种开始。2016 年多国科学家参与的国际花生测序团队发表二倍体野生种A.duranensis(V14167)和A.ipaensis(K30076)的基因组序列[12];广东省农业科学院2016 年发表了二倍体野生种A.duranensis(PI475845)的基因组序列[11],并于2018 年发表了二倍体野生种A.ipaensis(ICG8206)的基因组序列[10];河南农业大学2018 年发表了野生四倍体花生A.monticola(PI263393)的基因组序列[20]。2019 年花生栽培种3 个基因组先后发表:广东省农业科学院发表了四倍体栽培花生伏花生的全基因组序列,组装基因组大小为2.55 Gb,包含83 087 个蛋白编码基因;福建农林大学主导完成四倍体栽培花生狮头企的全基因组序列,组装染色体水平基因组大小约2.54 Gb,包含83 709 个蛋白编码基因;美国发表了四倍体栽培花生Tifrunner 的基因组序列,组装基因组大小约2.55 Gb,包含66 469 个蛋白编码基因。上述4 个物种8 个基因组的测序结果统计如表1 所示。高质量参考基因组为花生分子标记开发、基因定位与挖掘、全基因组水平的分子辅助育种等奠定重要的序列基础,促进花生分子遗传改良。我国花生研究者在花生基因组测序方面的研究成果大大提升了我国花生基础研究的国际地位。
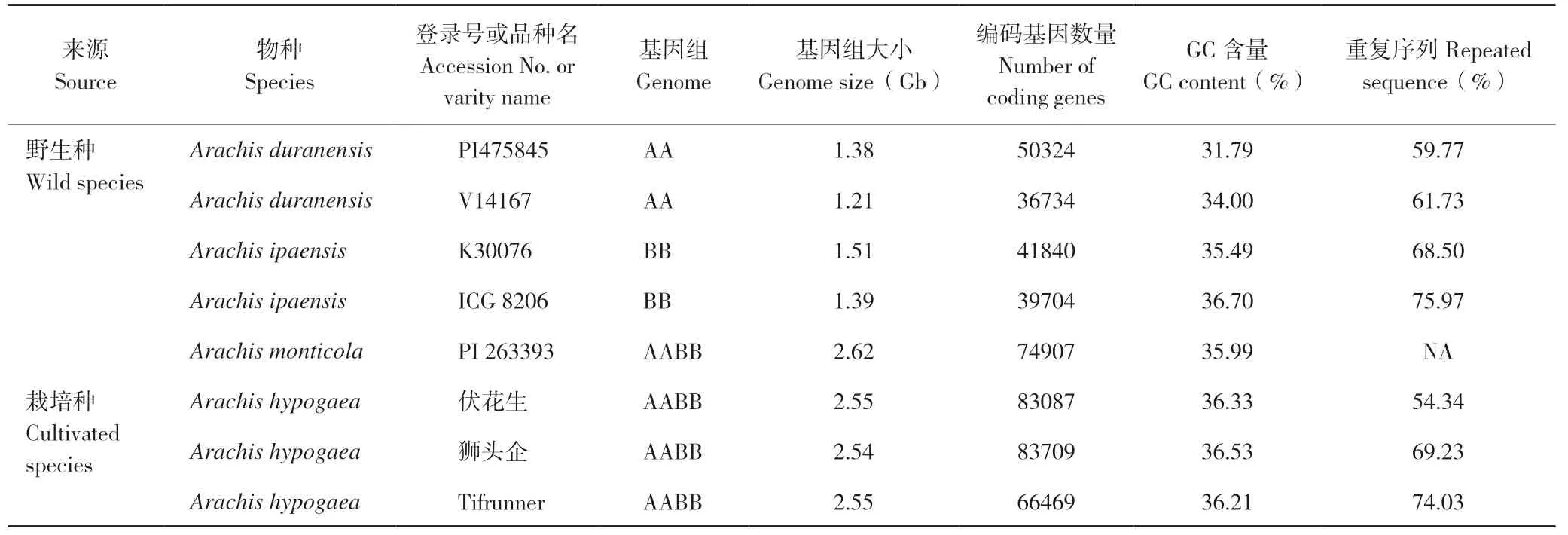
表1 花生野生种和栽培种全基因组测序结果Table 1 Results of genome sequencing of wild and cultivated peanut species
1.3 花生分子标记开发
分子标记在分子育种中发挥了重要作用,花生中已开发利用了多种分子标记,包括限制性内切酶片段长度多态性(Restriction Fragment Length Polymorphism,RFLP)[21]、随机扩增多态性DNA(Randomly Amplified Polymorphic DNA,RAPD)[22]、扩增片段长度多态性(Amplified Fragment Length Polymorphism,AFLP)[23]、序列特征性扩增区域(Sequence Characterized Amplified Regions,SCAR)[24]、切割扩增多态性序列(Cleaved Amplified Polymorphic Sequence,CAPS)[25]、多样性序列芯片技术(Diversity Array Technology,DArT)[26]、竞争性等位基因特 异 性PCR(Kompetitive Allele Specific PCR,KASP)[27]、简单序列重复(Simple Sequence Repeat,SSR)[28]、单核苷酸多态性(Single Nucleotide Polymorphism,SNP)[29-30]以及小片段插入和缺失标记(Insertions and Deletions,InDels)[31]等,其中SSR 和SNP 标记是目前最常用的两种标记。
花生SSR 标记的开发主要通过以下几种方法:(1)构建富含SSR 的DNA 文库[32];(2)细菌人工染色体(Bacterial Artificial Chromosome,BAC)末端测序(End Sequencing,BES)[33];(3)基于基因表达序列标签(Expressed Sequence Tags,EST)序列[28];(4)基于基因组Survey 序列[34];(5)基于全基因组序列[35]。花生SSR的早期开发比较困难,Hopins 等最早于1999 年开发利用花生SSR[36]。He 等利用SSR 富集DNA文库鉴定了56 个SSR 位点,其中只有19 个具有多态性[32]。Wang 等利用BAC 末端测序方法从36 435 个BES 中鉴定了1 424 个SSR 位点[33]。随着花生EST 数量的增加和RNA-seq 的广泛应用,通过EST 序列开发的SSR 标记越来越多,标记数量也急剧增加。Liang 等利用24 283 条EST序列开发881 个SSR 位点,其中290 个可用于标记开发[28]。徐志军等基于RNA-Seq 数据,鉴定出19 143 个SSR 位点,其中13 477 个SSR 位点可用于分子标记开发。花生全基因组测序的完成促进SSR 的开发数量进一步提升。Zhao 等基于二倍体野生花生基因组为参考序列,分别从A.duranensis和A.ipaensis中鉴定出135 529 和199 957 个SSR 位点,其中51 354 和60 893 个SSR 位点可用于分子标记开发[37]。Lu 等利用伏花生参考基因组分别从A 和B 亚基因组中鉴定了3 772 653 和4 414 961 个SSR 位点,其中462 267和489 394 个SSR 位点可用于分子标记开发[35]。
SNP 具有突变频率较低、等位基因较少、遗传稳定性较高和较其他标记分布范围更广等特点,为目前使用较为广泛的分子标记之一。SNP 位点的开发主要采用全基因组重测序(Whole-Genome Resequencing,WGRS)和简化基因组测序,其中简化基因组测序技术主要包括基因分型测序技术(Genotypong-by-sequencing,GBS)、限制性酶切位点关联DNA 测序技术(Restriction-Site Associated DNA Sequencing,RADseq)、特异性位点扩增片段测序技术(Specific-Locus Amplified Fragment Sequencing SLAF-seq)。Alves 等从野生花生中开发与抗性基因相关的SNP 标记[30]。Hong 等通过序列扩增方法开发了52 个SNP 位点,包括18个EST-SNP 和44 个genomic-SNP[29]。Zhang 等基于SLAF-seq 方法开发17 338 个花生SNP[38]。Pandey 等通过对41 个不同基因型花生材料重测序分别在A 基因组和B 基因组中鉴定了98 375 和65 407 个SNP 位点,通过进一步验证从中筛选出58 233 个SNP 并开发高密度SNP 芯片[39]。SNP 标记通过不同基因型材料的测序数据与参考基因组序列比较,获得覆盖全基因组的高密度标记位点,有利于开展高分辨率的性状关联分析。
1.4 花生遗传图谱构建
随着不同类型花生分子标记的大量开发,基于单一或整合多类型标记的花生遗传图谱相继发表。早期花生遗传图谱的构建主要基于RFLP、RAPD 和AFLP 等低密度传统分子标记[40-42]。随着SSR 标记的开发利用,近年来花生遗传图谱的构建主要采用SSR 标记,而且标记数量不断增加。SSR 标记刚开始用于遗传图谱构建时所含标记数量较少。Hong 等以142 个重组自交系(Recombinant Inbred Line,RIL)群体为材料,构建了包含131个SSR 标记的花生遗传图谱[43]。随后Varshney等也构建了一张包含135 个SSR 标记的遗传图谱[44]。Wang 等通过BES 测序开发4 152 个SSR引物,鉴定了385 个多态性位点,构建一张包含318 个SSR 标记的遗传连锁图谱,总遗传距离为1 674.4 cM,平均遗传距离为5.3 cM[33]。由于SSR 在栽培花生中的多态性低,基于一个RIL群体构建的遗传图谱所含标记数量较少,为构建高密度的遗传图谱,出现了整合多个群体的遗传图谱。Hong 等整合3 个RIL 群体构建了一张包含175 个SSR 标记的遗传图谱,涵盖22 个连锁群,总遗传距离为885.4 cM[45]。Sujay 等以两个RIL 群体为材料,构建了一张包含225 个SSR 标记的遗传图谱,总遗传距离为1 152.9 cM[46]。Gautami 等构建了3 个RILs 群体的整合图谱,包含293 个标记位点,分布在20 个连锁群,覆盖2 840.8 cM[47]。Gautami 基于11 个作图群体,构建高密度的整合遗传图谱,包含895 个SSR 标记和2 个CAPS 标记,涵盖20 个连锁群,总遗传距离为3 863.6 cM[48]。在此基础上,Shirasawa 通过整合另外5 个作图群体构建了一张更高密度的遗传图谱,标记数量达3 693,并能够将20 个连锁群定位到相应的花生A 和B 亚基因组[49]。
随着大量SNP 的开发,SNP 标记也逐渐应用于花生遗传图谱构建。Zhou 等利用RADseq 技术开发SNP 位点,以此构建一张包含1 621 个SNP标记和64 个SSR 标记的花生栽培种遗传图谱,涵盖20 个连锁群,总遗传距离为1 446.7 cM[50]。Agarwal 等利用WGRS 开发SNP 标记并构建涵盖20 个连锁群,包括8 869 个SNP 标记的高密度遗传图谱,总遗传距离为3 120 cM,平均遗传距离为1.45 cM[51]。Khan 等构建了两张分别包含1 975 个SNP 标记和5 022 个SNP 标记的高密度遗传连锁图谱用于抗黄曲霉相关基因定位和候选基因鉴定[52]。de Blas 等以栽培种和人工合成的双二倍体野生花生杂交的RIL 群体为材料,构建了一张包含1 819 个SNP 标记的遗传图谱,涵盖21个连锁群,总遗传距离为2 531.81 cM[53]。
2 花生传统育种技术及其局限性
2.1 表型鉴定与分析
表型分型或性状调查是育种选择的重要依据。无论是传统育种还是分子育种方法,都需要准确可靠的表型数据。育种者需要从成千上万的植株中筛选出综合性状优良的后代,如何鉴定表型、利用表型数据选择优良性状并淘汰不利性状,是传统育种选择的关键[54]。
表型鉴定与分析的方法涉及到植物生理、植物病理、生物化学以及食品加工等方面的原理和方法,根据调查性状的不同而有所差异。产量是品种选育必需的调查性状,产量性状分析可以通过百果重或小区产量作为衡量指标。抗性分析(如黄曲霉、青枯病、茎腐病等)可以在温室或实验室通过人工接菌,以及田间人工接菌或病圃中自然发病进行抗性分析[55-56]。品种的熟期可以用积温(Cumulative Thermal Time,CTT)来衡量。品质和营养成分通常采用化学方法检测,但是化学方法会破坏种子,影响后续选择,无损检测技术如近红外光谱分析(Near Infrared Reflectance Spectroscopy,NIRS)更适用于油脂、蛋白、碳水化合物或者脂肪酸等品质成分的检测。
表型鉴定分析有田间数据也有室内实验数据,如由黄曲霉菌引起的病害,既需要田间收获前的抗性调查数据,也需要收获后实验室的种子侵染调查数据。田间数据一般需要多年多点的重复数据,实验室数据一般需要3 个以上的重复。花生杂交之后从F2代开始出现分离,对目标性状的选择可以从F2代开始,有些杂交组合的分离群体比较大,而且F2代的种子需要进一步繁殖后代进行筛选,因此需要高通量、低成本、无损表型鉴定方法。
过去几十年传统育种技术在花生新品种选育方面取得显著成效,但是传统育种主要依赖于表型鉴定分析,存在随机性大和效率低的问题,随着表型组学发展,未来有望通过严格控制生长条件,开展大规模表型精准鉴定。
2.2 遗传变异与育种
与野生花生相比,栽培花生的遗传变异少,栽培种与野生种之间存在生殖隔离,阻碍了野生花生遗传变异向栽培花生的转移。除远缘杂交外,花生育种亲本通常只有保存的栽培花生种质资源或育种产生的高世代材料。在亲本数量有限的情况下,明确性状的遗传变异规律对于培育具有目标性状的品种非常重要。研究表明,锈病[57]、根结线虫[58]、油酸与亚油酸比值(油亚比,O/L)[59]等是由一对或少数几对主基因控制的质量性状,晚斑病抗性[60]、种子休眠性[61]具有数量遗传特性,另外,耐旱性和种子大小具有加性遗传变异的数量性状[62-63]。但是由于缺乏合适有效的表型分析方法,还有很多复杂农艺性状的遗传规律不清。
花生是闭花授粉的自交作物,适宜自花授粉作物的育种方法均可在花生育种中应用,包括群体选择、系谱法、集团选择、单粒传和回交选择方法等。目标性状在亲本之间的差异及其遗传力是育种亲本选择的重要参考因素。花生中应用的育种技术有传统杂交技术、远缘杂交、物理或化学诱变、细胞工程、转基因、分子标记辅助以及多技术聚合育种等[5]。随着基因分型成本越来越低,基于全基因组水平的分子育种技术应用也越来越广泛[64]。
3 基因组学在花生育种中的应用
3.1 性状连锁与关联分析
表型与基因型的关联分析是开展分子育种的重要前提,随着花生基因组资源的丰富,花生产量、品质、抗病、抗逆等重要性状的定位与关联研究均取得了较大进展[65]。在耐旱性状方面,Gautami 等定位了153 个主效QTL 和25 个上位性QTL[47]。Ravi 鉴定了52 个与耐旱相关的主效QTL,并发现这些QTL 与2~9 个性状相关联,表明耐旱性状的复杂性与数量性状特征[66]。在晚斑病抗性方面,Sujay 等鉴定了28 个晚斑病抗性QTL 和15 个锈病抗性QTL 位点,其中晚斑病抗性主效QTL 位点可以解释10.27%~62.4%的表型变异(Phenotypic Variation Explained,PVE),锈病抗性QTL 位点的PVE 可达82.67%[46]。Luo 等利用QTL-seq 分析在B 亚基因组第2 染色体上鉴定了两个抗青枯病QTL 位点[67]。Luo 等鉴定了33 个茎腐病抗性QTL 位点,其中6 个位点可解释10%以上的表型变异[68]。Pandey 等鉴定了78个主效QTL 和10 个上位性QTL 与花生含油量及油脂品质相关[69]。基于RIL 群体构建遗传图谱开展性状连锁分析所涉及的性状少,而利用自然群体开展的全基因关联分析可同时分析几十个性状,但也需更大的群体。Pandey 等基于300 份花生资源50 个重要农艺、病害和品质相关性状的全基因组关联分析,鉴定了36 个性状与524 个标记关联,对表型变异的解释介于5.81%~90.09%[70]。
3.2 分子标记辅助选择育种
标记辅助选择(Marker Assisted Selection,MAS)育种利用遗传标记将部分功能验证的候选标记与性状关联,在早期通过基因型分析进行选择,缩短世代间隔,加快育种进程。通过分子标记辅助可以使传统育种方法6~8 年的育种周期缩短到2~3 年。近10 年来,随着花生基因组学的发展,基因组资源极大丰富,为分子育种提供重要理论与数据支撑。分子标记辅助主要采用标记辅助的回交选择(Marker-Assisted Backcrossing,MABC)和标记辅助轮回选择(Marker-Assisted Recurrent Selection,MARS)方法。MABC 方法已成功应用于线虫抗性和高油亚比[71]。Cavanagh以抗锈病材料为轮回亲本,通过3 次回交和1 次自交,利用1 个显性和3 个共显性的标记,将锈病抗性主效QTL 引入其他农艺性状优良的感锈病花生材料,获得抗锈病的基因渗入系(Introgressed Lines,IL)[72]。国际半干旱热带作物研究所鉴定了1 个对锈病抗性贡献率达82.96%的主效QTL 位点,利用分子标记辅助选择技术成功培育抗锈病和叶斑病的高产花生品种[73]。同时,通过分子标记辅助整合高油酸、抗锈病和叶斑病的花生新种质[74]。Janila 等利用FAD2基因开展分子标记辅助选择,培育系列高油酸花生材料[75]。徐平丽等利用TaqMan 探针标记与KASP 标记结合的分子辅助轮回亲本选择创制了高油酸花生材料[76]。高油酸分子辅助育种已经取得较大进展[77-78]。
3.3 基因组选择
基于性状连锁的分子标记辅助选择适用于单基因或几个主效基因控制的性状,对于多基因控制的复杂性状比较困难。GS 是一种利用覆盖全基因组的高密度标记进行选育的新方法,尤其对低遗传力、难测定的复杂性状具有较好的预测效果,能快速准确地选择复杂性状[79-80]。GS 通过覆盖全基因组范围内的高密度标记进行育种值估计,获得不同染色体片段或单个标记效应值,然后将个体全基因组范围内片段或标记效应值累加,获得基因组估计育种值(Genomic-Estimated Breeding Values,GEBV)[79]。利用GS 进行育种选择,需要构建训练群体(Training Population,TP)建立相应的育种模型,然后通过育种群体(Breeding Population,BP)或候选群体(Candidate Population,CP)进行育种选择。采用育种骨干亲本材料建立TP,其中的每个材料都有已知的基因型和表型,通过适当的统计学建模,建立GS育种模型,估计每一个标记的效应值,然后利用同样的分子标记测定育种群体的基因型,根据标记效应值预测育种群体个体GEBV,最后根据GEBV 排名对个体进行选择[81]。GS 预测的准确性受TP 大小与组成结构、表型分型准确性、标记密度以及性状遗传力等因素影响[80]。
随着花生全基因组测序的完成、高通量测序技术的发展、基因分型成本的急剧下降,开展基于全基因组水平的GS 育种成为可能。国际半干旱热带作物研究所采用DArT 标记和SNP 芯片对花生微核心种质的开花天数、种子重量和荚果产量开展GS 模型构建和分析[39,82]。基因组选择的优势在于其对后代选择是基于对整个基因组而不是基因组中某一个或几个片段,真正实现了基因组在育种中的应用,将成为未来花生育种的重要育种方法和技术变革方向[80,83]。
4 展望
花生基因组学日新月异、蓬勃发展,产生了海量基因组资源,这些基础数据资源只有在育种中被充分利用转化,真正进入田间和市场才能体现基因组资源的价值和意义。目前,花生基因组学在育种中应用需要更多特异性群体。虽然已经有很多群体被构建并应用于花生遗传图谱构建、新材料创制和新品种选育,但是花生基因组学基础研究和育种实践仍需要更多特异性群体,如目标性状单一、群体数量更大的RIL 群体、多亲本高世代互交系(Multi-Parents Advanced Generation Intecross,MAGIC)[72]和染色体代换系(Chromosome Segment Substitution Lines,CSSL)等[84]。此外,花生基因组学在育种中应用需要高通量精准表型鉴定技术。获得准确和精细表型数据是开展花生全基因组选择的关键[85]。随着表型组学发展,通过自动、遥感、多场景数据整合实现高通量、准确、精准表型鉴定是未来数字化育种的重要发展方向[80]。全基因组水平的选择育种是未来育种发展的必然趋势。基于单个性状连锁标记的分子辅助选择将逐渐发展为基于全基因组水平育种值估测的全基因组选择[86],基于测序或基因分型的全基因组水平的育种选择将在未来花生育种中占据主导地位[64]。
