Theoretical study of(e,2e)triple differential cross sections of pyrimidine and tetrahydrofurfuryl alcohol molecules using multi-center distorted-wave method
2022-01-23YiaoWang王亦傲ZhenpengWang王振鹏MaomaoGong宫毛毛ChunkaiXu徐春凯andXiangjunChen陈向军
Yiao Wang(王亦傲), Zhenpeng Wang(王振鹏), Maomao Gong(宫毛毛),Chunkai Xu(徐春凯), and Xiangjun Chen(陈向军)
Hefei National Laboratory for Physical Sciences at Microscale and Department of Modern Physics,University of Science and Technology of China,Hefei 230026,China
Keywords: (e,2e),bio-molecules,multi-center distorted-wave method(MCDW)
1. Introduction
During the past decades, human genome has become an increasing concern globally, especially for the radiation damage process when exposing to the severe environment.The single-strand breaks, double-strand breaks, DNA-protein cross-links,base release and other chemical modifications can cause the genome damage.[1]Plenty of experimental and theoretical advances have been focus on the direct and indirect processes of low-energy electrons on DNA damage, which can lead to loss of genetic information, mutation, promotion of genomic instability, and apoptosis.[2-5]The damage occurs directly to the individual DNA moieties or by the indirect reaction with the species around DNA.Researchers spent much effort on the subject, trying to understand the interaction between the low-energy electrons and DNA bases, water, sugar analogs and so on. In order to describe the biological effects, many groups have developed Monte Carlo track structure codes to simulate the charged-particles paths in living tissues.[6-8]Cross section data of molecules will be used in the Monte Carlo codes. But due to the difficulty of experimental measurement,the theoretical molecular scattering data for complex bio-molecules are desired.
Pyrimidine (C4H4N2) is an important bio-molecule,which possesses a six-membered ring structure with two nitrogen atoms locating in the 1 and 3 positions. It is a prototypical structure for the DNA nucleobases of cytosine(C4H5N3O), thymine (C5H6N2O2), as well as RNA base uracil(C4H4N2O2). And it is naturally considered as a model compound to investigate the electron collision with DNA and RNA bases.
Tetrahydrofurfuryl alcohol(THFA,C5H10O2)is regarded as a basic unit of a phosphate deoxyribose backbone which is the major structural component in DNA. It contains a five-member heterocyclic furanose ring, which undergoes pseudo-rotation[9]that may produce 20 possible conformers in the gaseous THFA.[10]The molecular structure of THFA in gas phase has been studied by the electron diffraction experiment[10]andab initiotheoretical calculation.[10]Together with pyrimidine, the two bio-molecules are two typical model compounds of nucleobases and phosphate deoxyribose backbone to investigate electron collisions with DNA constituents,respectively.
Measurements on triple differential cross sections (TDCSs)of electron impact single ionization have been carried out for a series of bio-molecules in the last twenty years, such as tetrahydrofuran (THF, C4H8O),[11,12]tetrahydropyran (THP,C5H10O),[12]pyrimidine,[13]tetrahydrofurfuryl alcohol,[9,14]and thymine (C5H6N2O2).[15]Due to the anisotropic multicenter nature and the complex electronic structure of the molecular system, there are only a few theoretical calculations for this process. Madison and coworkers[16-22]developed a molecular three-body distorted-wave(M3DW)model.It is usually not satisfactory when it is used to explain the cross section of bio-molecules, because the orientation averaged molecular orbital (OAMO) approximation neglects the multi-center nature of the complex target. In order to be more precise,OAMO approximation was replaced by the proper average(PA)[23-26]to retrieve the multi-center nature in the initial state. Other calculations based on the Coulomb wave and simplified distorted wave models[27-30]were also reported in recent years.
A multi-center distorted-wave method (MCDW) was developed[31-33]in our group to calculate the (e, 2e) cross sections of molecules at relatively higher impact-energy under asymmetric kinematics,where the energy of the scattered electron is much higher than the ejected one. The incident and scattered electrons are approximately described by plane waves due to the high impact-energy,while the wave function of the ejected electron is calculated in the anisotropic potential of molecular ion. Through this method, the influence of the molecular multi-center nature can be largely included. Then TDCS is obtained in the framework of first Born approximation(FBA)by averaging over all molecular orientations. The MCDW method not only has been applied to the TDCS calculations of inorganic small molecules,[31,33,34]but also acts as a good model for some bio-molecules.[35,36]
In the present work,MCDW method is applied to calculate the TDCSs of pyrimidine and THFA molecules in coplanar asymmetric kinematics. The experimental works were reported in the literature. Present MCDW calculations well reproduced the experimental data.The paper is organized as follows.In Section 2,we briefly outline the MCDW method.The results and comparisons with experimental data and other calculations as well as discussions will be presented in Section 3,followed by the summary in Section 4. Atomic units are used throughout the paper unless explicitly stated otherwise.
2. Theoretical method
Since the details of the MCDW method have been given in Refs.[31-33],here we will only briefly outline its formulation. The MCDW method is developed within the framework of FBA under asymmetric kinematics. In the scattering theory,the transition amplitude for a given molecular orientation in laboratory reference reads

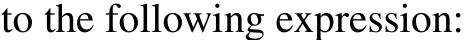

whereK=ki-ksis the momentum transfer.Rnis the position of then-th nucleus, andZnindicates its charge. Vectorrerepresents the position of the active electron.|F(-)〉is the continuum wave function of the ejected electron, and|φα〉is the bound orbital to be ionized. The first term in Eq.(2)represents the scattering by the active electron,and the second term refers to the scattering by the nuclei. In the present MCDWNT calculation,the nuclear term will be fully included.[32,33]
The ejected electron is regarded as moving in the anisotropic field of the residual ion,while the interaction from the fast scattering electron is neglected. The continuum wave function of the ejected electron can be obtained by solving the effective Schr¨odinger equation

The modeling of such interaction is rather difficult because its nature depends on the distance of the incoming electron,varying from a purely polarization effect at large distances up to including the exchange-correlation interaction with the bound electrons within the molecular volume. The spatial factorsrcare obtained from the crossing between the lower polarization potential terms and the ones given by the correlation potential.
The anisotropic multi-center feature ofF(-)is inherited fromVm. To solve this equation, the single-centered expansion technique[37-39]is employed, where the wave function and potential are expanded over the symmetry-adapted angular functions. Note that the model potentialVmis anisotropic and introduces the coupling between terms of different angular momentum in the partial wave expansion ofF(-), resulting in a set of coupled equations. As shown in our previous work,[31-33]the diagonal terms in the potential matrix are considered dominant.Thus in practical calculation,we will ignore the off-diagonal elements and solve the decoupled partial wave equations.
The TDCS is then obtained by averaging over all possible molecular orientations,)
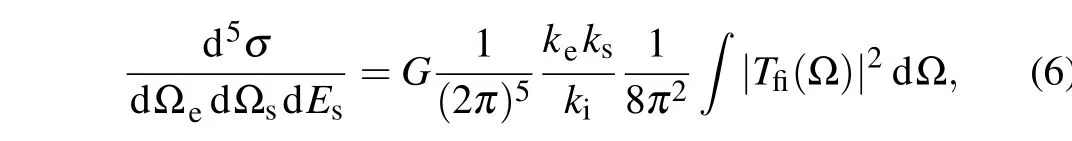
whereGis the electron occupation number of the ionized molecular orbital.
3. Results and discussion
In the present calculations, the geometries of pyrimidine and THFA molecules are optimized at MP2[40-44]/augcc-pVTZ[45]level using Gaussian 09 program.[46]The bound wave functions of the molecules are obtained by density functional theory employing the B3LYP hybrid functional[47,48]and aug-cc-pVTZ basis set. The bound orbital and the continuum wave function are both expanded over the symmetryadapted angular functions with up limits oflbmax= 8 andlcmax= 18 in the calculation. The convergence is reached with radial range from 0 to 8.47 a.u., and Euler angle meshNα=Nβ=Nγ=30, whereNα,Nβ, andNγare the numbers of points for Euler anglesα,β,andγ,respectively.
3.1. Pyrimidine
The experiment of pyrimidine[13]was carried out in the coplanar asymmetric kinematics atEi= 250 eV andEe=20 eV.The reported TDCS experimental data includes highest occupied molecular orbital(HOMO)7b2and outer valence orbital 10a1. Due to the limited experimental energy resolution,the HOMO cannot be fully resolved from the next HOMO 2b1,and the 10a1,1b1and 6b2orbitals are recognized as one peak in the deconvoluted process. The previous theoretical calculations of M3DW model only consider the contribution of 7b2and 10a1orbitals.[13]Here, a more complete description using MCDW method is presented in Figs. 1 and 2. The electron density maps of the corresponding bound orbitals are also plotted in the figures.
In Fig.1,the TDCS of the combined orbitals(7b2+2b1)is displayed at kinematic condition ofEi= 250 eV,Ee=20 eV, andθs=-15°, including experimental data and theoretical results of MCDW-NT method. The distribution of TDCS exhibits two lobes structure, the binary lobe is around the momentum transfer direction(K),which is generally recognized as the binary (e, 2e) collisions between the incident and bound electrons. The other is around the opposite direction(-K),called recoil lobe,which is resulted from the backward reflection of the ejected electron in the molecular ion potential after the collision. Actually, the kinematic condition is not far from the Bethe-ridge, the binary peak will partially reflect the momentum profile[29]of the ionized bound orbital,which has been investigated previously in the literature. The splitting of the binary lobe of the experimental data can be well described by MCDW-NT, but cannot be reproduced by M3DW-OAMO model in the literature.[13]The shape of the TDCS is also similar with the calculated result of Mouawadetal.[29]by averaging over all molecular orientations, as indicated by the green solid line(there are no absolute cross sections in the original paper,[29]so we normalize the result of DW model with the best visual fit to experiment). It reflects the importance of TDCS contributions of different molecular orientations in the initial state. In the experimental work,[13]7b2and 2b1orbitals are deconvoluted by two Gaussian peaks.However,due to the limited energy resolution,the two orbitals cannot be resolved from each other. So the calculation results of 7b2and 2b1orbitals are also plotted for comparison. It can be seen that the total cross sections of 7b2and 2b1orbitals are better than the result of each individual orbital when comparing with the experiment.

Fig. 1. The TDCSs for ionization of the combined 7b2+2b1 orbitals of pyrimidine molecule in coplanar geometry. The incident and ejected electron energies are 250 eV and 20 eV,respectively. The scattering angle and the momentum transfers are -15° and |K|=1.12 a.u. The experimental data (solid square points) is normalized to the result of MCDW-NT theory(red solid line) with best visual fit. The calculation results of MCDW-NT theory for individual 7b2(magenta dotted curve)and 2b1(blue dotted curve)orbitals are also plotted for comparison. The green solid line indicates the result of DW model based on FBA.[29]
In Fig. 2, the TDCS of the combined outer valence orbitals(10a1+1b1+6b2)by MCDW-NT method is displayed at kinematic condition ofEi= 250 eV,Ee= 20 eV andθs=-5°,-10°,-15°. For different scattering angles, fromθs=-5°toθs=-15°,the kinematic condition gradually approaches the Bethe-ridge. The single binary lobe structure changes to a weak splitting double peak, as indicated by the red solid line. The TDCSs of each individual states are also plotted for comparison. In Fig.2(c),the weak splitting double peak of the binary lobe can be well described by MCDW-NT method, but overestimates the cross section at ejection angle around 110°. A same result can be observed in Fig.2(b). The present calculation model fails to predict the recoil lobe at all the conditions. It mainly comes from the multi-center effect when the ejected electron moves in the ionized ion potential,which cannot be fully included at present calculation stage.The DW model[29]also achieves good agreements with experiment,and is present for comparison.tion gradually approaches to the Bethe-ridge.From Figs.3(a)-3(c),the single broad binary peak changes to oscillatory structure in the binary region. The oscillatory structure actually indicates the s-p type momentum profile,as have been investigated in the electron momentum spectroscopy.[9]The MCDW calculation well predicts the shape of experimental data in the binary region, especially forθs=-15°. The TDCS of each individual conformer is also plotted in the figure. The contribution of conformer B (16±8%) can be neglected in the total TDCS. The recoil lobe of experimental data still cannot be described by MCDW-NT method,which is the same as all the kinematic conditions of pyrimidine molecule in Figs.1 and 2. The large error bars of experimental data,especially in Fig.3(b),make it difficult to evaluate the theoretical model.

Fig.2. The TDCSs for ionization of the combined 10a1+1b1+6b2 orbitals of pyrimidine molecule in coplanar geometry. The incident and ejected electron energies are 250 eV and 20 eV, respectively. The scattering angle and the corresponding momentum transfers are (a) -5°, |K|=0.47 a.u.;(b) -10°, |K|=0.78 a.u.; (c) -15°, |K|=1.12 a.u. The experimental data (solid square points) is normalized to the result of MCDW-NT theory(red solid line) with best visual fit. The calculation results of MCDW-NT theory for individual 10a1 (magenta dotted curve), 1b1 (blue dotted curve)and 6b2 (red dotted curve) orbitals are also plotted for comparison. The green solid line indicates the result of DW model.[29]

Fig. 3. The TDCSs for ionization of the HOMO of tetrahydrofurfuryl alcohol molecule in coplanar geometry. The incident and ejected electron energies are 250 eV and 20 eV, respectively. The scattering angle and the corresponding momentum transfers are(a)-5°,|K|=0.45 a.u.;(b)-10°,|K|=0.77 a.u.; (c) -15°, |K|=1.12 a.u. The experimental data (solid square points) is normalized to the result of MCDW-NT theory (red solid line) with best visual fit. The calculation results of MCDW-NT theory for conformer A(magenta dotted curve)and B(blue dotted curve)are also plotted for comparison.
3.2. THFA
Compared to pyrimidine molecule,the structure of THFA molecule is more complicated. Twenty conformers of THFA may be produced through pseudo-rotation in gaseous phase.[9]There are two main conformers with abundance of 84±8% and 16±8%[14]through the investigation by electron diffraction[14]andab initiomethods.[14]For the convenience,we mark the two conformers as A(84±8%)and B(16±8%),respectively.
Figure 3 shows the TDCS results of HOMO(28a)ionized THFA molecule atEi=250 eV,Ee=20 eV andθs=-5°,-10°,-15°. Similar with Fig.2,with the increase of the scattering angle fromθs=-5°toθs=-15°,the kinematic condi-
In Figs. 4 and 5, we also present calculations based on the Coulomb wave(CW)model to compare with MCDW-NT model. In the CW model,the continuum wave function of the ejected electron is simply described as the Coulomb wave,instead of the multi-center continuum wave function in MCDWNT method. The results of CW model show similar distributions with MCDW-NT model both in the binary and recoil regions. It shows that the short range multi-center potential of pyrimidine molecular ion is not so important to predict the TDCSs, which is different from the reported results of THP and 1,4-dioxane molecules.[36]In Fig. 6, the comparison between CW and MCDW-NT models for THFA molecule is displayed. Unlike pyrimidine molecule,the result of CW model is quite different from that of MCDW-NT model in the binary region. It reflects that the multi-center effect for different molecular target on the ejected electron will have different impact on the cross sections. The similar calculation results of CW and MCDW-NT methods for pyrimidine molecule are probably because of its high molecular symmetry.

Fig.5.The comparison of MCDW-NT and CW methods of the combined 10a1+1b1+6b2 orbitals for pyrimidine molecule with the same kinematic condition in Fig.2.

Fig. 4. The comparison of MCDW-NT and CW methods of the combined 7b2+2b1 orbitals for pyrimidine molecule with the same kinematic condition in Fig.1.
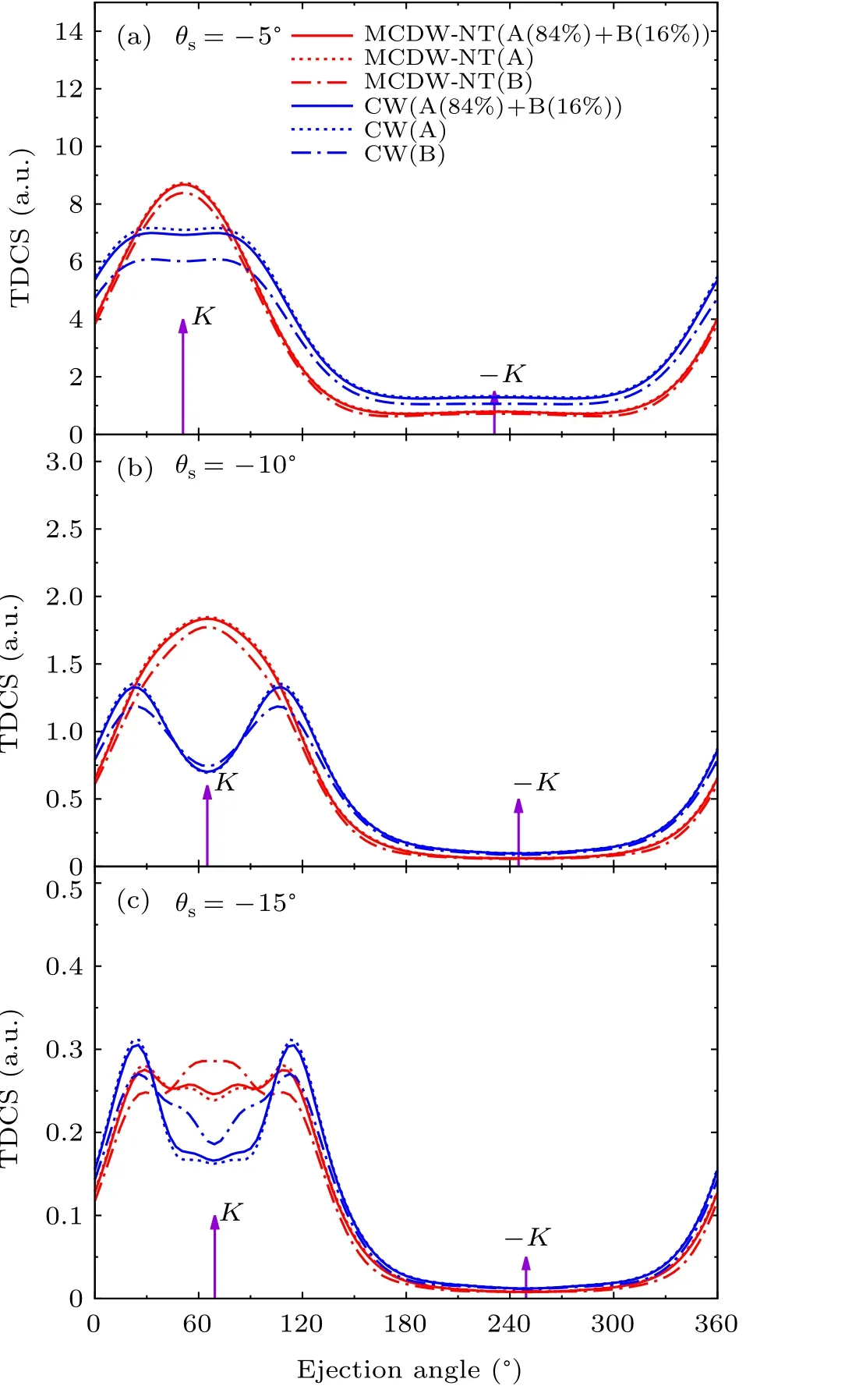
Fig.6.The comparison of MCDW-NT and CW methods of the HOMO for tetrahydrofurfuryl alcohol molecule with the same kinematic condition in Fig.3.
From the TDCS calculations of pyrimidine and THFA molecules, model compounds of nucleobases and phosphate deoxyribose backbone, the MCDW behaves as a good theoretical model to investigate electron collisions with DNA constituents,especially in the binary region. However,the model has limitations when the more accurate calculated results are required. The high-order effect beyond FBA and the strong coupled effect between low energy electron and multi-center ion potential are still two main problems. More accurate theoretical models and experimental data are desired in the future.
4. Summary
In this paper, a theoretical study of electron impact ionized TDCSs of two bio-molecules, the model compounds of nucleobases and phosphate deoxyribose backbone in DNA constituents,pyrimidine and THFA,is reported. Good agreements are achieved between MCDW method and experimental data atEi=250 eV,Ee=20 eV in the coplanar asymmetric kinematic condition, especially in the binary region. MCDW method fails to predict the recoil lobe of experimental data.It may come from the high-order effect beyond FBA and the strong coupled effect between low energy electron and multicenter ion potential. For such bio-molecules,the precision of(e,2e)cross section is a long-standing challenge,as the multicenter nature has greatly increased the computational complexity and quantity. More accurate theoretical models and experimental data are desired in the future.
Acknowledgments
Project supported by the National Natural Science Foundation of China (Grant Nos. 12004370, 11534011, and 11934004) and the National Key Research and Development Program of China (Grant Nos. 2017YFA0402300 and 2019YFA0210004).
杂志排行

Chinese Physics B的其它文章
- Superconductivity in octagraphene
- Soliton molecules and asymmetric solitons of the extended Lax equation via velocity resonance
- Protection of entanglement between two V-atoms in a multi-cavity coupling system
- Semi-quantum private comparison protocol of size relation with d-dimensional GHZ states
- Probing the magnetization switching with in-plane magnetic anisotropy through field-modified magnetoresistance measurement
- Majorana zero modes,unconventional real–complex transition,and mobility edges in a one-dimensional non-Hermitian quasi-periodic lattice