结合多尺度卷积胶囊网络的植物lncRNA编码小肽预测
2022-01-22胡鹤还赵思远纪腾其
胡鹤还, 孟 军, 赵思远, 纪腾其
(大连理工大学 计算机科学与技术学院 辽宁 大连 116023)
0 引言
长非编码RNA(iong non-coding RNA, lncRNA)是长度大于200个核苷酸(nt)的非编码RNA,在植物生长、发育等进程中发挥作用[1]。从是否编码蛋白质的角度,lncRNA起初被认为不具备编码能力而归结为非编码RNA(non-coding RNA, ncRNA)。然而,最近研究表明,部分lncRNA中含有不超过300 nt的短开放阅读框(short open reading frames, sORFs),具备编码小肽的能力。这些lncRNA能够在细胞质中与核糖体结合。核糖体在lncRNA上不断移动,对长度不超过300 nt的sORFs进行翻译,从而形成一类长度小于100个氨基酸(amino acid, aa)的小肽。这类由lncRNA的sORFs编码小肽(sORFs-encoded small peptides, SEPs)在植物生命活动中发挥了调节作用[2]。sORFs以及SEPs的发现,使得mRNA和ncRNA的界限变得模糊(部分ncRNA同样具有编码能力),同时有助于提升人们对基因组学的整体认知。
目前识别SEPs的方法主要分为生物实验方法和计算方法两类。生物实验方法一方面造价高、耗时长,另一方面不适用于大规模的鉴定。计算方法大多基于人类和动物数据训练的机器学习模型,考虑到动植物ncRNA之间由于生成过程中聚合酶的不同而存在差异[3],因此,植物SEPs与动物SEPs之间可能同样存在一定的差异,传统机器学习模型涉及过多的人工干预,未充分挖掘SEPs存在的深层特征。所以,采用深度学习方法挖掘植物SEPs,已成为发展趋势。
本文采用生物信息学软件sORF finder和ORF finder挖掘植物lncRNA中的sORFs,使用基因组学中生物序列的连续编码方式,对sORFs序列进行编码后作为模型输入,提出了一种结合多尺度卷积胶囊网络(capsule network, CapsNet)的深度学习模型。该模型兼顾了卷积层充分提取初级局部特征与CapsNet提取高级特征并自动进行特征聚类的特性,使两者达到互补,从而更好地实现对lncRNA中sORFs的分类预测。通过与单一、简单融合的深度学习模型的比较,以及对多个物种数据集的测试,结果表明本文提出的模型具有良好的分类效果和泛化能力。
本文的主要贡献如下:
1) 采用生物信息学软件挖掘植物lncRNA中的sORFs,将植物lncRNA编码小肽的预测问题转化为判断sORF是否具有编码潜力的问题。
2) 基于逻辑推理的思想,将生物信息学软件挖掘出的sORFs进一步筛选,提升数据的可信度。
3) 提出了一种结合多尺度卷积胶囊网络的深度学习模型来实现植物lncRNA编码小肽的预测。
1 相关工作
目前,对SEPs的研究大多采用生物实验和计算方法。生物实验方法主要有核糖体检测、质谱分析、肽组学分析等[4],研究人员采用上述生物学实验方法,鉴定出了一系列SEPs。随着研究的深入,已有研究者采用上述方法对SEPs进行了大规模分析。Fesenko等[5]针对小立碗藓以及其他10种植物(拟南芥、玉米和卷柏等),通过质谱分析技术得到具有高可信度的SEPs。然而,生物实验耗费大量人力物力,不适合进行大规模的SEPs鉴定。
与生物实验方法相比,计算方法节省了大量的时间和成本。识别sORFs和SEPs的生物信息学工具主要使用机器学习方法。例如,Hanada等[6]基于编码序列(CDS)与非编码序列(NCDS)之间的六聚体组成偏差,采用perl语言编写了适用于拟南芥等11种生物的工具包sORF finder。Zhu等[7]开发了一种专门用于鉴定SEPs的基于机器学习方法的工具MiPepid,该工具通过提取人类氨基酸序列对应RNA序列,以及人类非编码RNA中sORFs序列的4-mer特征,并使用逻辑回归模型提出了工具MiPEPid。Tong等[8]分别提取了人类和动物的小编码RNA和小非编码RNA的序列与理化特征,并依托支持向量机(SVM)构建了工具CPPred。Zhang等[9]使用了与CPPred相同的数据集,提出了一种基于CNN的RNA编码潜力预测工具DeepCPP,表明深度学习方法挖掘SEPs已逐步进入公众视野。然而,已有的生物信息学工具大多挖掘人类和动物SEPs,由于动植物SEPs之间可能存在一定的差异,因此目前的工具不能挖掘具有高可信度的植物SEPs。
2017年Sabour等[10]在NIPS会议中提出了胶囊网络(CapsNet)结构。CapsNet采用了一种新的“向量进,向量出”的传递方案,使用向量代替标量表示对应特征,从而使得输出向量的每个维度代表“特征的特征”。CapsNet结构有两个重要的创新:Squash压缩激活函数和动态路由。Squash压缩激活函数将向量的模长压缩到0~1之间,从而使得向量模长能够代表对应特征的概率;动态路由通过强化相似特征、弱化离群特征,从而做到特征聚类。
为了增加特征多样性,减少关键信息的损失,本文结合CNN与CapsNet的优势,提出了一种结合多尺度卷积胶囊网络的深度学习模型,用三种不同尺度的卷积核分别提取特征,并将提取结果分别转化成三个不同尺度的胶囊矩阵。多尺度卷积胶囊网络不仅能够更加充分地提取特征,还能够考虑sORFs序列中数据之间的相关性,从而更好地实现lncRNA编码小肽的预测,并为相应的生物学实验打下了基础。
2 数据预处理
2.1 数据集构建
从GreeNC数据库(http:∥greenc.sequentiabiotech.com/wiki/Main_Page)下载拟南芥(Arabidopsisthaliana)、大豆(Glycinemax)以及苔藓(Physcomitrellapatens)的lncRNA数据。分别使用生物信息学软件sORF finder(http:∥hanadb01.bio.kyutech.ac.jp/sORFfinder/)、ORF finder[11](https:∥www.ncbi.nlm.nih.gov/orffinder/)获取lncRNA的sORFs。ORF finder是一个图形的序列分析工具,能够分析并找到序列中的sORFs。sORF finder基于编码序列(CDS)与非编码序列(NCDS)之间的六聚体组成偏差,进而识别序列中具有编码能力的sORFs。取两种工具识别结果的交集与差集,通过CD-HIT[12]本地化工具,以0.8作为阈值,进行去冗余处理,得到候选sORFs序列,其中两种工具结果的交集作为正集,差集作为负集。获取候选sORFs的流程如图1所示。

图1 候选sORFs的获取流程
2.2 序列编码
对于候选的sORFs序列,采用p-nts编码方式[13],将每p个连续的碱基作为一个子序列,子序列间不重叠。由于连续三个相邻的碱基构成一个密码子,所以本文的p取3,即对正、负样本中所有序列进行分词处理后,统计得出一个大小为43=64的生物单词表。按照单词在生物序列中出现的频率,从大到小进行编码。由于sORFs的长度不超过300,所以可将sORFs序列嵌入到一个100维向量中。例如,当输入序列S=(GAGGCCGTT……ACTCTATGT)时,根据上述编码方式,每三个连续碱基视为一个单词,再按词频大小进行编码,即可将S编码视为一个固定长度的向量SC=(8,55,11,…,37,58,14)。向量SC为模型的最终输入格式。
3 特征分析与逻辑推理
3.1 特征分析
为了探究CDS与候选正集,NCDS中sORFs与候选负集在序列组成和理化特性方面存在的差异性,针对sORFs序列和氨基酸序列分别采用不同的特征编码方式进行分析。对于sORFs序列,提取其k-mer特征[14];对于氨基酸序列[15],分别提取其188D、双氨基酸组成(di-peptide composition,DPC)特征。然后基于奇异值分解(singular value decomposition, SVD)、主成分分析(principal component analysis, PCA)、t分布随机相邻嵌入(t-distributed stochastic neighbor embedding, t-SNE)、核主成分分析(kernel principal component analysis, KPCA)四种特征降维方法,将特征降为二维。上述3类特征编码方式具体如下。
1)k-mer特征描述序列组成信息,每个k-mer为序列中相邻的连续k个核苷酸。本文设定k为4,共256维,其中每一维度代表对应k-mer的频率fk-mer:
(1)
2) 188D特征综合考虑了氨基酸组成(amino acid composition,AAC)特征,氨基酸类别组成过滤和分布特征(composition transition distribution,CTD),共188维。其中:前20维描述20种氨基酸的组成频率fAAC;后168维描述8大类氨基酸的理化特性fCTD,

(2)
3) DPC特征描述了氨基酸序列中双核苷酸的组成信息,共400维,其中每一维度代表对应双核苷酸的频率fDPC,
(3)
3.2 逻辑推理
本文采用的逻辑推理框架如图2所示。

图2 逻辑推理结构
反绎学习[16]由机器学习和逻辑推理两部分构成。给定一组数据,机器学习部分中的初始分类器给出伪标签,导致伪事实,然后基于最小化与知识库的不一致性,通过逻辑推理修正伪标签,替换原分类器,重复迭代直到伪标签与知识库完全一致为止。本文将候选数据集的正负标签视为伪标签,将CDS、NCDS中sORFs的特征视为知识库,对候选正负集进行筛选。具体做法如下。
1) 将候选正集与CDS的特征、候选负集与NCDS中sORFs的特征进行比对。
2) 获取CDS、NCDS中sORFs的特征的横、纵坐标范围,然后将其视为知识库,筛选掉不在CDS特征坐标范围内的候选正集、不在NCDS特征坐标范围内的候选负集。
4 多尺度卷积胶囊网络模型
4.1 嵌入阶段
嵌入层的作用是将输入序列映射成卷积层易于处理的矩阵向量的形式,方便卷积层充分提取特征。主要工作是将输入序列的每个数字映射成一个1×n维的向量,这样输入序列被映射成m×n维的矩阵形式,其中:m代表序列长度;n代表嵌入维度。本文中序列长度m为100,嵌入维度n为64,即嵌入阶段每条序列可映射为100×64的矩阵,作为多尺度卷积胶囊阶段的输入。嵌入层使用Keras库的Embedding()方法,参数output_dim设置为64,input_length设置为100。
4.2 多尺度卷积胶囊阶段
卷积神经网络(convolutional neural networks, CNN)是深度学习中一种被广泛应用的网络[17],主要由卷积层、池化层、全连接层构成。卷积层采用卷积核沿着矩阵向量以指定滑动步长逐个进行卷积计算,具体计算公式为
(4)

由于CNN中的池化层会导致关键信息的丢失,同时也会忽略整体与部分之间的关联,因此采用胶囊网络代替池化层。研究表明,模型性能与特征多样性呈正相关,即特征种类越丰富,模型性能越好。由于固定尺度的卷积核只能提取某一局部特征,难以捕捉到不同局部特征,从而忽略某些潜在信息。为克服这一缺陷,引入多尺度卷积核和多个胶囊层代替单尺度卷积核和单一胶囊,从而避免特征提取不充分导致有效信息丢失。实验中采用三个不同尺度的卷积核对嵌入层的输出分别进行卷积操作,每个卷积操作可捕捉序列不同位置的局部特征,然后对每个卷积操作的特征映射矩阵,采用胶囊网络将其转换为三个胶囊矩阵,最后将胶囊矩阵进行串联,作为多尺度卷积胶囊层的输出。
本文的卷积核尺寸分别为3×64、6×64、9×64,即卷积层的输出维度分别为98×64、95×64、92×64;然后将每个特征映射矩阵分别转化为8、12、16个胶囊,每个胶囊的维度为16,即胶囊矩阵的维度分别为8×16、12×16、16×16;最后采用Concatenate()函数将所有胶囊矩阵串联,形成108×16的胶囊矩阵作为次级胶囊层的输入。
4.3 分类阶段
胶囊之间通过动态路由算法进行连接,低一级胶囊转换成更高一级的胶囊,从而自动进行特征聚类,更好地表达高级特征。其具体实现如图3所示。

图3 动态路由算法流程图

(5)
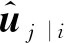
(6)

(7)
利用Squash非线性激活函数处理输入向量sj,得到高级胶囊层的输出向量vj,
(8)
(9)
动态路由机制是一个迭代算法,当迭代次数等于指定值时迭代终止,即bij停止更新。此时得到的vj即为高级胶囊层的最终输出向量。
本文将多尺度卷积胶囊阶段生成的108×16的胶囊矩阵视为108个16维胶囊,基于动态路由算法将其转换成10个16维胶囊,即10×16的胶囊矩阵。利用Flatten层处理该胶囊矩阵得到160维向量,再添加参数为0.4的Dropout层防止过拟合,最后使用参数为1的Dense层得出一个具体数字,并使用sigmoid()函数将其映射在[0,1]之间,即得出预测标签。
4.4 模型实现
输入序列(Input)经过p-nts编码(Coded)形成100维向量。首先利用嵌入层(Embedding)将输入向量映射成一个100×64的矩阵向量,方便进行卷积操作;然后通过多尺度卷积胶囊层(Multi-scale Convolution and CapsNet)进行操作,输出108×16的胶囊矩阵,再基于动态路由算法(Dynamic Routing)形成10×16的胶囊矩阵,最后经分类得出预测结果,模型的整体结构如图4所示。

图4 模型整体结构
5 实验与结果
5.1 验证方法与评价指标
采用苔藓数据作为训练集,拟南芥和大豆作为独立测试集。通过与现有深度学习方法比较,验证提出的模型在sORFs编码小肽预测方面的性能与泛化能力。
实验采用5折交叉验证法来验证模型的性能。使用准确率(Accuracy,ACC)、精确率(Precision,P)、召回率(Recall,R)以及F1值(F1_score,F1)对模型性能进行评估。评价指标的定义如下。
1) 准确率(ACC)表示被正确预测的样本所占比例,
(10)
2) 召回率(R)表示实际的正样本在被正确预测的样本中所占比例,
(11)
3) 精确率(P)表示被预测为正的样本中,实际的正集样本所占比,
(12)
4)F1值(F1)为召回率与精确率的加权平均值,
(13)
其中:TP表示正确分类的正样本;FN表示被分为负样本的正样本;TN表示正确分类的负样本;FP表示被分为正样本的负样本。
5.2 数据集
从GreeNC数据库中下载相关数据并进行数据预处理,然后通过逻辑推理过程筛选数据集。为保证正、负样本均衡,从负集样本库中随机抽取与正集相同数目的样本作为负集。各物种具体数据如表1所示。

表1 各物种数据集
5.3 基于不同方法的分类结果
为验证提出方法的有效性和优势,实验采用苔藓数据集,按照p-nts编码方式对sORFs序列进行编码,并使用卷积神经网络(CNN)、长短期记忆网络(Bi-LSTM)、胶囊网络(CapsNet)、CNN+CapsNet、CNN+Bi-LSTM方法进行对比实验,5折交叉验证的实验结果如表2。表中性能最好的用粗体表示。
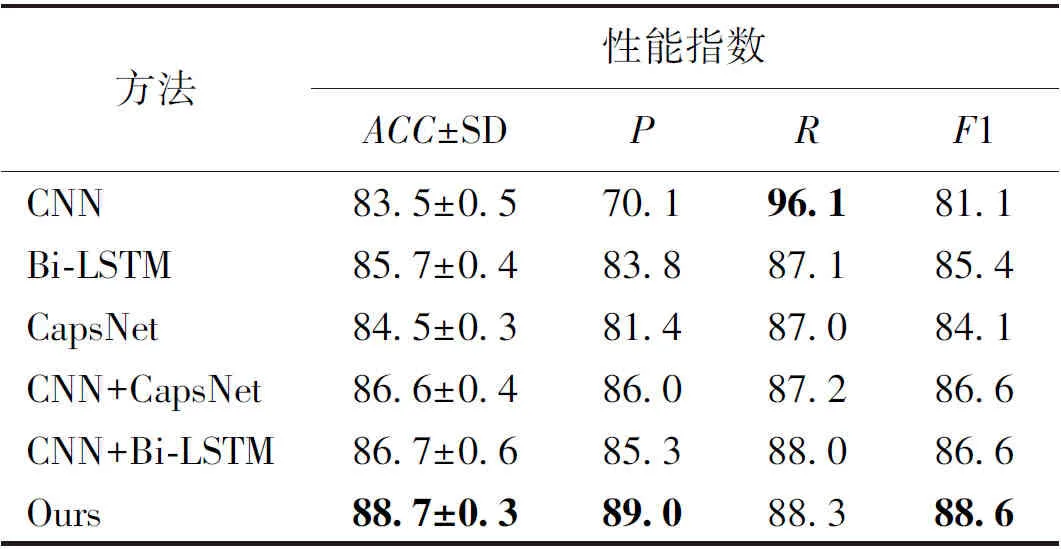
表2 基于不同方法的分类结果
从表2可看出,较之单一深度学习方法,提出方法在准确率、精确率、召回率和F1值4个指标性能上都具有明显的优势。在准确率上比CNN、Bi-LSTM和CapsNet分别提升了5.2%、3.0%、4.2%,说明提出方法在预测sORFs是否具有编码能力方面具有良好的分类能力;同时,与单尺度模型CNN+CapsNet相比,提出模型能够提取不同局部特征,充分挖掘潜在信息;与CNN+Bi-LSTM模型相比,提出的模型既能提取丰富的特征,又能自动进行特征聚类,输出更具有表达力的特征向量。在准确率上比CNN+CapsNet、CNN+Bi-LSTM分别提升了2.1%、2.0%。
5.4 基于不同物种的分类结果
为证明提出方法的泛化能力,选用拟南芥、大豆数据集作为独立测试集,使用CNN+CapsNet、CNN+Bi-LSTM方法进行对比实验。独立测试的实验结果如表3。表中性能最好的用粗体表示。从表3结果可看出,在拟南芥、大豆两个物种的独立测试集上,提出方法的性能与CNN+CapsNet和CNN+Bi-LSTM方法相比都有一定的提升。提出方法在预测拟南芥、大豆的sORFs是否具有编码能力的性能较好,表明模型具有良好泛化能力。

表3 基于不同物种的分类结果
6 结束语
本文提出一种多尺度卷积胶囊网络的深度学习模型,使用多尺度卷积核捕捉不同局部特征,然后使用多层胶囊网络捕捉深层次特征并自动进行特征聚类,从而实现lncRNA编码小肽的分类预测。实验结果表明,提出模型与传统深度学习和单尺度模型对比,取得了最好的分类效果。此外,模型在多物种独立测试集上也取得了良好的分类效果,验证了提出模型具有健壮的泛化能力。未来将对植物lncRNA编码小肽开展更加深入细致的研究,如直接使用模型对输入的植物lncRNA进行分析,识别出其中的编码小肽区域,进而进行生物学验证。
